Aze A and Maiorano D. Recent advances in understanding DNA replication: cell type–specific adaptation of the DNA replication program [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1351 (https://doi.org/10.12688/f1000research.15408.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
DNA replication is an essential process occurring prior to cell division. Cell division coupled to proliferation ensures the growth and renewal of a large variety of specialized cell types generated during embryonic development. Changes in the DNA replication program occur during development. Embryonic undifferentiated cells show a high replication rate and fast proliferation, whereas more differentiated cells are characterized by reduced DNA synthesis and a low proliferation rate. Hence, the DNA replication program must adapt to the specific features of cells committed to different fates. Recent findings on DNA synthesis regulation in different cell types open new perspectives for developing efficient and more adapted therapies to treat various diseases such as genetic diseases and cancer. This review will put the emphasis on recent progress made in this field.
Keywords
DNA synthesis, nucleus, chromatin, epigenetics, development, cell cycle, differentiation
Corresponding author:
Domenico Maiorano
Competing interests:
No competing interests were disclosed.
Grant information:
Research in DM’s laboratory is supported by grants from “Fondation ARC pour la Recherche sur le Cancer”, La Ligue contre le Cancer, INSERM, and MSD Avenir.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
DNA synthesis occurs during the S phase of the cell cycle and is ensured by the replisome, a molecular machine made of a large number of proteins acting in a coordinated manner to synthesize DNA at many genomic locations, the replication origins1. Replication origin activation in space and time (or replication program) is set by a sequence of events, starting already at the end of mitosis, lasting through G1 phase, and ending in S phase when DNA replication is activated. These coordinated events ensure that the full genome will be replicated before mitosis since a faithful DNA synthesis is a prerequisite for genome integrity maintenance2. DNA replication initiates from thousands of replication origins scattered along the chromosomes. Origins acquire the competence to replicate in a step called “licensing” that involves formation of a pre-replication complex (pre-RC), including loading of the replicative helicase MCM2-7 (recently reviewed 3). Then once the transition from pre-RC to a pre-initiation complex (pre-IC) is induced by S-phase kinases, DNA replication is activated and DNA is unwound to provide the template for the replicative DNA polymerases2,4.
The last 30 years of deep investigations in the DNA replication field have allowed the general mechanisms involved in eukaryotic DNA synthesis to be defined. Thanks to recent methodological improvements, such as genome-wide analysis5,6, in vitro reconstitution assays7–9, proteomics10–12, and structural biology13,14, our vision of the molecular pathways that govern DNA synthesis is becoming sharper.
Although the factors that drive DNA replication initiation are well conserved throughout the eukaryotic kingdom, cell type–specific differences in the regulation of this process within eukaryotes have been recently unraveled. From these studies, it appears that the regulation of DNA replication is influenced by the fate of a given cell. These findings shed new light on how the DNA replication program and the proliferation rate are being modulated during cell fate commitment, when cell type specialization is determined. New discoveries in this topic are undoubtedly essential to expand our understanding about the genesis of diseases and their progression. In this review, we will briefly describe recent achievements aiming to understand how the replication factors involved in the licensing, activation, and elongation steps of DNA synthesis adapt dynamically with cell fate determination to maintain genome integrity and homeostasis.
Coordination between replication origin licensing and G1 length is critical for cell destiny
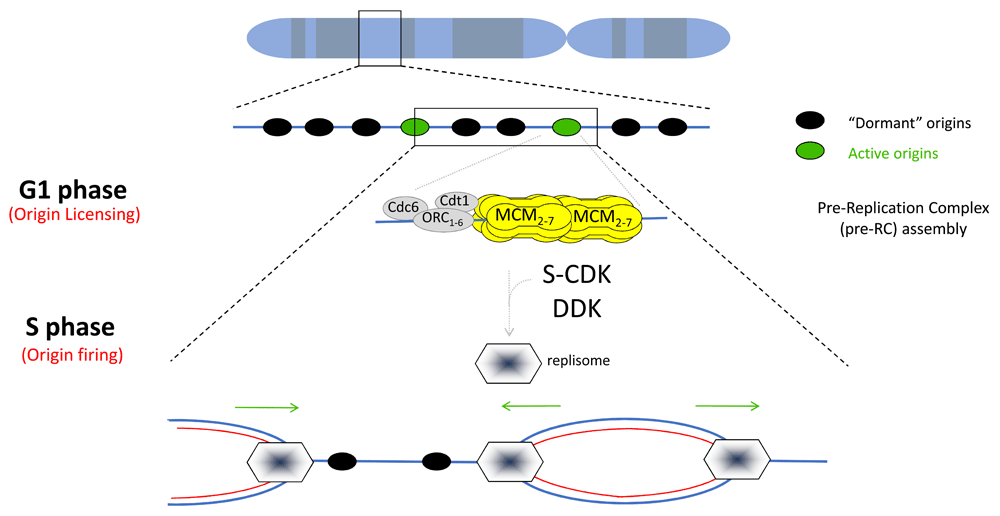
In vitro reconstitution assays in yeast have provided tremendous detailed information on how origins are licensed by dissecting the sequential biochemical steps involved3,7–9,14,15. Detailed reviews covering the main features that characterize metazoan origins have been recently published3,5,16. Briefly, in eukaryotes, replication origins are licensed through a chronological order that requires the binding of Origin Recognition Complex (ORC), Cdc6, and Cdt1 proteins onto chromatin17,18. Two hexamers of the MCM2-7 helicase complex then are loaded in an inactive state prior to S phase (Figure 1). In yeast and mouse cells, Cdt1 and MCM2-7 form a complex before being recruited onto replication origins by ORC and Cdc619–21. The MCM2-7 double hexamer must be activated by S-phase kinases to be able to unwind DNA and initiate DNA replication. However, MCM2-7 complexes are recruited in excess so that not all MCM2-7 complexes are activated during a cell cycle (Figure 1). The choice of the origins to be activated (around 30,000 in mammalian cells) is variable from cell to cell and ensures flexibility in origin usage during the DNA replication program to adapt with cell fate commitment, cell environment, and replicative stress22,23. Origin mapping strategies coupled with high-throughput sequencing revealed that the chromatin context influences origin selection through genetic and epigenetic features located in close proximity to the origins.
Figure 1. Replication of eukaryotic chromosomes.
Replication origins are scattered along the genome to ensure that each chromosome is entirely replicated in S phase. Pre-replication complex (Pre-RC) assembly and origin activation are tightly regulated in a sequential manner to ensure that replication occurs only once per cell cycle. Hence, throughout the G1 phase, origins are licensed by the sequential loading of the pre-RC components: Origin Recognition Complex (ORC), CDC6, CDT1, and (at the final stage) MCM2-7. Once a double MCM2-7 hexamer is stably recruited onto chromatin, origins are licensed. Many replication origins are licensed but few are activated, allowing a backup of origins to be used when DNA replication is perturbed. Origin firing occurs under the combined activities of S-phase Cyclin-Dependent Kinase (CDK) and Dbf4-Dependent Kinase (DDK), allowing the recruitment of additional factors involved in DNA replication initiation and elongation. MCM2-7, together with CDC45 and GINS, composes the DNA helicase that unwinds DNA in a bidirectional manner. The entire replication machinery made of accessory factors required for replication fork stability and the synthesis of DNA is called the replisome.
It is now clear that most replication origins being constitutively activated across multiple metazoan cell types (hereafter named “shared origins”) are associated with unmethylated CpG islands, G-rich elements, transcriptional start sites, and histone modifications related to open chromatin marks (H3K4me3, H3K9Ac, and H3K27Ac)24–29. Notably, this population of shared origins tends to initiate early during S phase, whereas cell type–specific origins initiate during late S-phase and are linked with compacted chromatin marks28. These observations underline the relationship between chromatin modifications with the cellular context in origin selection.
Two independent studies describing reconstitution of DNA replication initiation events in vitro from chromatinized templates in yeast provided biochemical evidence that the regulatory functions of chromatin structure influence origin selection. These studies confirmed that nucleosome-free regions contribute to defining ORC binding and thus origin function30,31, as previously suggested from genome-wide studies mapping ORC binding sites and nucleosome occupancy in various organisms32–36. Moreover, in mouse embryonic stem (ES) cells, depletion of the histone H1 perturbs the landscape of replication origin activation37. Because chromatin environment changes during cell fate commitment and in different cell types, it is generally assumed that the DNA replication program is coordinated with transcription to avoid transcription–replication conflicts and thus preserve genomic integrity. This seems particularly crucial during the onset of developmental programs and cell lineage specification. In Caenorhabditis elegans, origins from rapidly replicating pluripotent embryos coincide with open chromatin regions of highly transcribed genes, whereas establishment of the new transcriptional program, occurring at later embryonic stages when cell differentiation begins, correlates with the reorganization of replication initiation sites29,38.
Permissive chromatin features for rapid activation of replication origins could influence the cell cycle length. ES cells have a shorter G1 compared with their differentiated counterparts, which impacts on the licensing step. ES cells recruit much more MCM2-7 to the chromatin than tissue-specific stem cells or progenitor cells39. This excess of origins that remain dormant during S phase appears to be important to maintain pluripotency40,41 and could also protect the genome against DNA replication stress occurring in ES cells39,42.
Besides changes in the kinetics by which MCM2-7 is loaded onto chromatin, licensing control adapts to G1 length, suggesting that pre-RC binding is developmentally regulated. Indeed, quantitative single-cell analysis performed in human cells by Matson and colleagues demonstrated that origins are licensed faster in pluripotent cells compared with their isogenic differentiated counterparts and that loading of the MCM2-7 helicase slows down as G1 duration is extended43. The authors revealed that high expression of the licensing factor Cdt1 was important for fast MCM2-7 loading rates, thus enabling ES cells to rapidly license origins prior to the G1-to-S phase transition. Similar to the need for additional dormant origins, fast licensing kinetics seems essential to ensure pluripotency in ES cells and induced pluripotent stem cells. This observation was corroborated by Carroll and colleagues in intestinal stem cells from adult tissues44. The authors demonstrated that licensing is interconnected with the proliferative commitment of stem cells. They found that Lgr5+ intestinal stem cells, which contribute primarily to the renewal of the intestinal epithelium, reside mostly in a G1, unlicensed state, although they express MCM2 and other proliferative markers to a level equivalent to the licensed population. Interestingly, this unlicensed state correlates with an elongated cell cycle that could be considered as a backup mechanism to sustain proliferative fate decisions and tissue maintenance. Nonetheless, the link between chromatin structures and adaptation to G1 length in such a context remains to be determined.
Uncoupling of G1 length with licensing kinetics has been observed during oncogenic transformation. Overexpression of oncogenes, such as cyclin E and c-MYC, shortens G1 and forces cells to engage S phase earlier with incomplete licensing. Consequently, the replication program is perturbed, leading to accumulation of replication stress45,46. Origin usage following oncogene activation was recently investigated genome-wide. The authors found that in these conditions new initiation zones, which were normally suppressed by transcription in G1, appear in intragenic regions47. These results also confirm an observation in drosophila showing that active transcription modulates MCM2-7 distribution48. Nonetheless, how RNA polymerases inactivate and redistribute MCM2-7 remains to be determined. It has been reported that DNA translocases and RNA polymerases in yeast are able to push MCM2-7 along DNA, but similar processes have not yet been described in metazoans8,49. An emerging picture from these studies is that cell cycle length, local chromatin structure, and active transcription can modulate the extent of origin licensing.
Activation of DNA replication in different cellular contexts: control in space and time matters
During S phase, origins are activated by the combined activities of cyclin-dependent kinase (CDK) and Dbf4-dependent kinase (DDK) kinases (Figure 1) targeting several substrates allowing the recruitment of CDC45 and GINS complex to MCM2-7, thereby forming the CMG complex, the functional replicative helicase50. Chromatin environment contributes to licensing as described previously through MCM2-7 loading but also to origin selection through MCM2-7 activation51,52. The dynamics of origin activation during S phase follow a spatio-temporal order known as replication timing (Figure 2). Replication timing in eukaryotes controls activation of replication of large chromosomal domains and is mediated by genetic determinants, local histone modifications, and global chromatin organization53. Thus, DNA sequence features that mediate licensing in vertebrates can also affect the time when origins will be activated. Origin G-rich repeated elements (OGREs) are prone to form G-quadruplexes (G4s). Their presence in the genome is associated with origin activity25,54. Nucleosome organization at the proximity of origins was reported to modulate both origin licensing (as described above) and MCM2-7 helicase-dependent activation steps of initiation55 in yeast and more recently in mammals37. Different histone modifications and certain histone modifiers have been assigned in origin selection and the dynamics of their activation across S phase29,38,52,56–60.
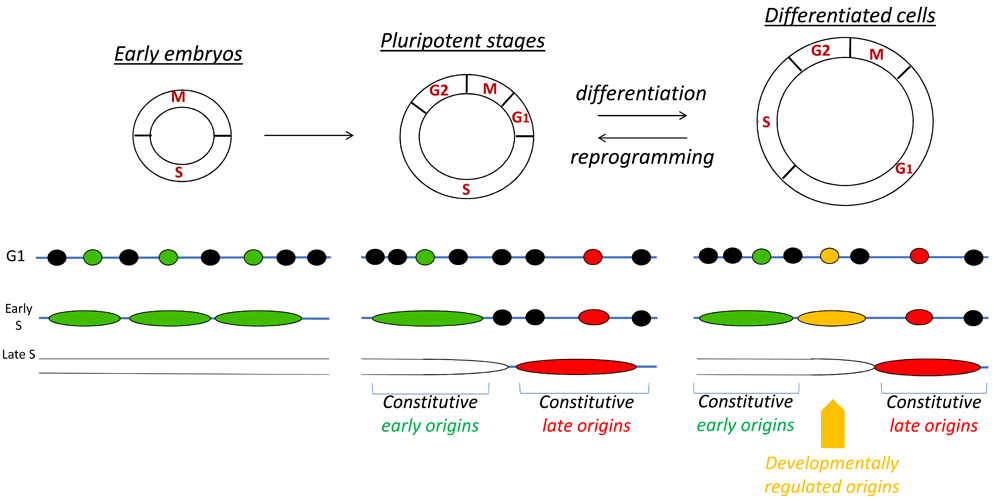
Figure 2. Replication program and cell features.
Changes in the DNA replication program are dependent on cell features. In those rapidly dividing embryos, which are transcriptionally inactive, many replication origins fire to ensure fast S-phase. Selection of replication origins at this stage is believed to be random. Despite improvements in replication origin mapping, this idea has not yet been challenged. The timing of replication during early development is not clearly defined, as S-phase length is very short. During the pluripotency stage, appearance of gap phases and the onset of transcription confer a certain nuclear organization that sets the genome for a specific program of replication. Constitutive origins that replicate early are associated with strong origin density and efficiency, a high GC content, strong gene density, and nucleosome-free regions. On the other side, late constitutive origins are poor in origins and genes, GC content is low, and chromatin accessibility is restricted. Rif1 is a major factor shown to modulate origin activity for late replicating domains. The replication program changes dynamically during differentiation or cell lineage development, in coordination with changes in transcriptional activity and chromatin organization. Whereas constitutive origins are activated at the same location and with the same timing, some origins will be regulated following acquisitions of new cellular features. Remarkably, tumor development leads to formation of a heterogeneous population of cells, including cells with stem-like properties, progenitors, and differentiated tumor cells. These cells have the ability to perpetuate their lineage, to give rise to differentiated cells, and to undergo rapid growth. These molecular features, recapitulated during early embryonic development, illustrate similarities between cancer stem cells and embryogenesis. Understanding the DNA replication program regulation in a cell type–specific manner will allow investigation of the processes behind tumorigenesis from a different perspective.
Several histone-modifying enzymes such as the methyltransferase PR-Set7 and the acetyltransferase HBO1 in complex with BRPF3 have been reported to have a role in activation of particular replication origins52,61. More recently, a great deal of investigation has focused on the contribution of chromatin regulators in controlling the timing of activation through chromatin organization. The telomere-associated protein Rif1 interferes with DDK-dependent phosphorylation of MCM2-7 and modulates nuclear architecture by anchoring heterochromatin62,63. Rif1 might organize replication-timing domains through its association with G4s to repress pre-RC activation until late S-phase63–65. The ORC-associated protein ORCA/LRWD1 establishes a repressive chromatin environment at a subset of origins, thus priming them for late replication66. The replication-initiation determinant protein RepID initiates replication in a sequence-specific manner by interacting with a subgroup of origins67.
Modulation of origin activation in a cell type–specific manner depends upon global changes in chromosomal architecture and mainly occurs within large chromosome domains known as Topologically Associating Domains (TADs)68–71. These constitute structurally distinct chromatin domains covering several megabases where DNA interactions are favored. TADs show a strong correlation with replication timing domains in which several replication units are concomitantly activated. Nuclear reorganization that accompanies differentiation and the establishment of developmental programs result in changes in replication timing of the human genome70,72,73; notably, alterations in replication timing have been detected in several diseases74,75.
Therefore, flexibility in origin activation seems critical to accommodate the dynamic changes in the transcriptional program and genome repositioning (Figure 2). Indeed, cell type–specific replication origins correlate with regions corresponding to differentiation-specific and tissue-specific gene expression programs76,77. Origins that are activated in a cell type–dependent manner appear to replicate late in S phase28. Histones and chromatin modifications described above impose a crosstalk between replication and transcription and are interconnected during differentiation and development29,38 although this relationship remains enigmatic.
Origin activation dynamics that determine replication timing therefore might play a role in establishing local and global chromatin structure to facilitate the cellular response to the differentiation process. Remarkably, establishment of TADs during early development in mammals requires DNA replication but not transcription78. Moreover, perturbations in DNA replication during development can cause epigenetic changes (alleviation of repressive marks) potentially inherited by the next generations79.
Chromatin organization immediately following fertilization in vertebrates is particularly critical for DNA replication initiation and this occurs in a transcription-independent manner following fertilization80.
The capacity to selectively modulate origin usage in a cell type–specific manner suggests that the proteins involved in origin activation might play specific functions. Indeed, replication initiation proteins, and their availability during S phase, can be involved in dictating specificity in origin activation81–85. The Sld3 vertebrate homolog Treslin/TICRR, a CDK target that acts as a binding site for TopBP1 and Mdm2 binding protein MTBP (both proteins being required for GINS-CDC45 recruitment), was proposed to link chromatin acetylation to DNA replication initiation efficiency and timing in different cancer cell lines58,86. This observation shows that Rif1 is not the only known site-specific regulator of DNA replication initiation and timing. Likewise, a function in origin efficiency has been assigned to the Treslin partner MTBP, probably through its ability to localize Treslin near G4 structures87. Finally, Rif1 accessibility to chromatin was recently implicated in determining the onset of late replication during embryonic development88.
Overall, these studies suggest a functional interaction between components of the replication machinery with chromatin modifiers leading to reorganization of the genome architecture during development. In turn, reorganization of the chromatin architecture defines cell type–specific transcriptional programs that feedback on the availability of replication proteins but equally on the replication timing by shaping chromatin architecture in specific domains such as TADs. Hence, genetic and epigenetic features set during developmental transitions may function as selective ways to repress or activate licensing or firing (or both) of a subset of origins, potentially within replication timing domains. Precise origin mapping at the single-molecule level in a single cell (using nanopore sequencing, for instance) completed by in vitro reconstitution assays using human proteins will undoubtedly bring to light new exciting concepts in this field.
Adaptation of the replisome to fast proliferation
A large variety of cell types critical for tissue function are formed during early embryonic development, when cell commitment first takes place. The proliferative state decision is variable for each cell type and may influence cell cycle duration. Consequently, steps required for proliferation, such as genome duplication, must be tightly regulated with cell cycle progression to maintain homeostasis. For instance, embryonic cell division in metazoans exhibits dramatic lengthening of the cell cycle at the onset of gastrulation, a stage required for cell type specialization and embryo patterning89,90. Lengthening of the total cell cycle time is achieved mostly by extension of the G1 phase and moderately the S phase91.
A variation in replisome composition following DNA replication perturbations or between different cell types was unraveled thanks to recent advances in quantitative proteomics at the replication forks11,92,93. Analysis of the replication machinery at the forks by isolation of proteins on nascent DNA (iPOND) allows comparison of protein abundance at replication forks in different contexts. It was shown that replisomes of pluripotent stem cells contain a particular protein network that accommodates a high proliferative capacity with short cell cycle phases and reduced endogenous DNA replication stress94. Factors involved in DNA repair, such as mismatch repair, but equally pluripotency and epigenetic inheritance factors, such as the NuRD-HDAC complex, were found to be enriched at replication forks. Interestingly, independent studies confirmed the requirement of a NuRD complex for DNA replication during early embryonic development95. ES cells seem to require additional factors at the replication forks to cope with DNA replication perturbations. Thus, Filia-Floped was identified by iPOND as a new protein complex involved in resolution of stalled forks during a normal S-phase in mouse ES cells compared with their differentiated counterparts96. It is likely that strategies set in stem cells to ensure fast DNA replication may also be exploited in other biological contexts requiring fast proliferation, including very early embryogenesis and tumorigenesis. For example, the Rad18 E3 ubiquitin ligase, a master regulator of translesion DNA synthesis, is an abundant component of the DNA replication machinery during Xenopus early development and confers to the replisome the ability to hijack DNA lesions and suppress the DNA damage response, ensuring fast cell cycle progression. Remarkably, Rad18 was found to be highly expressed in glioblastoma cancer stem cells97.
Overall, these recent observations converge toward the idea that replisome composition can be selected “a la carte” regarding the biological features set by specific cell types. Consistent with this idea, replication of specific genome locations like telomeres, common fragile sites, or centromeres has been shown to involve particular DNA replication partners at the vicinity of the DNA replication fork to ensure stability of those genomic regions98–100.
Conclusions
A defective DNA replication program is the source of several pathologies during development (Meier-Gorlin syndrome) and during the adult life (carcinogenesis)45,101. Metazoan origins lack consensus sequences and are associated with different structural features that confer cell type–specific replication. Additional mechanisms operate to fine-tune pre-RC assembly, origin activity, and replisome composition. This allows cells to coordinate a fast replication program with their fate, contributing to genome integrity maintenance despite DNA replication perturbations often observed during tumorigenesis and development. Identification of various factors involved in origin selection and their activity in a cell type–specific manner is a great source of interest. Such factors may often be deregulated during cancer development; thus, their targeting might constitute effective anti-cancer therapies.
Nevertheless, these discoveries are only the tip of the iceberg as limited information is currently available regarding their regulation. Biochemistry, electron cryomicroscopy analysis, in vitro reconstitution assays using vertebrate proteins, and genomic approaches on single cells should largely contribute to breakthrough findings in the field.
Abbreviations
CDK, cyclin-dependent kinase; DDK, Dbf4-dependent kinase; ES, embryonic stem; G4, G-quadruplex; iPOND, isolation of proteins on nascent DNA; ORC, origin recognition complex; pre-RC, pre-replication complex; TAD, topological associated domain
Grant information
Research in DM’s laboratory is supported by grants from “Fondation ARC pour la Recherche sur le Cancer”, La Ligue contre le Cancer, INSERM, and MSD Avenir.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank Philippe Pasero and Eric Morency for critical reading of the manuscript.
Faculty Opinions recommended
References
1.
Alabert C, Groth A:
Chromatin replication and epigenome maintenance.
Nat Rev Mol Cell Biol.
2012; 13(3): 153–67. PubMed Abstract
| Publisher Full Text
2.
Fragkos M, Ganier O, Coulombe P, et al.:
DNA replication origin activation in space and time.
Nat Rev Mol Cell Biol.
2015; 16(6): 360–74. PubMed Abstract
| Publisher Full Text
3.
Hyrien O:
How MCM loading and spreading specify eukaryotic DNA replication initiation sites [version 1; referees: 4 approved].
F1000Res.
2016; 5: pii: F1000 Faculty Rev-2063. PubMed Abstract
| Publisher Full Text
| Free Full Text
4.
Parker MW, Botchan MR, Berger JM:
Mechanisms and regulation of DNA replication initiation in eukaryotes.
Crit Rev Biochem Mol Biol.
2017; 52(2): 107–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
8.
Warner MD, Azmi IF, Kang S, et al.:
Replication origin-flanking roadblocks reveal origin-licensing dynamics and altered sequence dependence.
J Biol Chem.
2017; 292(52): 21417–30. PubMed Abstract
| Publisher Full Text
| Free Full Text
13.
Miller TC, Costa A:
The architecture and function of the chromatin replication machinery.
Curr Opin Struct Biol.
2017; 47: 9–16. PubMed Abstract
| Publisher Full Text
17.
Maiorano D, Moreau J, Méchali M:
XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis.
Nature.
2000; 404(6778): 622–5. PubMed Abstract
| Publisher Full Text
18.
Maiorano D, Rul W, Méchali M:
Cell cycle regulation of the licensing activity of Cdt1 in Xenopus laevis.
Exp Cell Res.
2004; 295(1): 138–49. PubMed Abstract
| Publisher Full Text
21.
You Z, Masai H:
Cdt1 forms a complex with the minichromosome maintenance protein (MCM) and activates its helicase activity.
J Biol Chem.
2008; 283(36): 24469–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Méchali M:
Eukaryotic DNA replication origins: Many choices for appropriate answers.
Nat Rev Mol Cell Biol.
2010; 11(10): 728–38. PubMed Abstract
| Publisher Full Text
23.
Yekezare M, Gómez-González B, Diffley JF:
Controlling DNA replication origins in response to DNA damage - inhibit globally, activate locally.
J Cell Sci.
2013; 126(Pt 6): 1297–306. PubMed Abstract
| Publisher Full Text
24.
Besnard E, Babled A, Lapasset L, et al.:
Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs.
Nat Struct Mol Biol.
2012; 19(8): 837–44. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
25.
Cayrou C, Ballester B, Peiffer I, et al.:
The chromatin environment shapes DNA replication origin organization and defines origin classes.
Genome Res.
2015; 25(12): 1873–85. PubMed Abstract
| Publisher Full Text
| Free Full Text
27.
Picard F, Cadoret JC, Audit B, et al.:
The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells.
PLoS Genet.
2014; 10(5): e1004282. PubMed Abstract
| Publisher Full Text
| Free Full Text
36.
Rodriguez J, Lee L, Lynch B, et al.:
Nucleosome occupancy as a novel chromatin parameter for replication origin functions.
Genome Res.
2017; 27(2): 269–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
39.
Ge XQ, Han J, Cheng EC, et al.:
Embryonic Stem Cells License a High Level of Dormant Origins to Protect the Genome against Replication Stress.
Stem Cell Reports.
2015; 5(2): 185–94. PubMed Abstract
| Publisher Full Text
| Free Full Text
42.
Ahuja AK, Jodkowska K, Teloni F, et al.:
A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells.
Nat Commun.
2016; 7: 10660. PubMed Abstract
| Publisher Full Text
| Free Full Text
45.
Macheret M, Halazonetis TD:
DNA replication stress as a hallmark of cancer.
Annu Rev Pathol.
2015; 10: 425–48. PubMed Abstract
| Publisher Full Text
46.
Yang Y, Gao Y, Mutter-Rottmayer L, et al.:
DNA repair factor RAD18 and DNA polymerase Polκ confer tolerance of oncogenic DNA replication stress.
J Cell Biol.
2017; 216(10): 3097–115. PubMed Abstract
| Publisher Full Text
| Free Full Text
54.
Cayrou C, Coulombe P, Vigneron A, et al.:
Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features.
Genome Res.
2011; 21(9): 1438–49. PubMed Abstract
| Publisher Full Text
| Free Full Text
56.
Liu J, McConnell K, Dixon M, et al.:
Analysis of model replication origins in Drosophila reveals new aspects of the chromatin landscape and its relationship to origin activity and the prereplicative complex.
Mol Biol Cell.
2012; 23(1): 200–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
57.
Rondinelli B, Schwerer H, Antonini E, et al.:
H3K4me3 demethylation by the histone demethylase KDM5C/JARID1C promotes DNA replication origin firing.
Nucleic Acids Res.
2015; 43(5): 2560–74. PubMed Abstract
| Publisher Full Text
| Free Full Text
64.
Kanoh Y, Matsumoto S, Fukatsu R, et al.:
Rif1 binds to G quadruplexes and suppresses replication over long distances.
Nat Struct Mol Biol.
2015; 22(11): 889–97. PubMed Abstract
| Publisher Full Text
65.
Mattarocci S, Shyian M, Lemmens L, et al.:
Rif1 controls DNA replication timing in yeast through the PP1 phosphatase Glc7.
Cell Rep.
2014; 7(1): 62–9. PubMed Abstract
| Publisher Full Text
68.
Gilbert DM, Takebayashi SI, Ryba T, et al.:
Space and time in the nucleus: Developmental control of replication timing and chromosome architecture.
Cold Spring Harb Symp Quant Biol.
2010; 75: 143–53. PubMed Abstract
| Publisher Full Text
69.
Pope BD, Hiratani I, Gilbert DM:
Domain-wide regulation of DNA replication timing during mammalian development.
Chromosome Res.
2010; 18(1): 127–36. PubMed Abstract
| Publisher Full Text
| Free Full Text
77.
Norio P, Kosiyatrakul S, Yang Q, et al.:
Progressive activation of DNA replication initiation in large domains of the immunoglobulin heavy chain locus during B cell development.
Mol Cell.
2005; 20(4): 575–87. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
80.
Aze A, Fragkos M, Bocquet S, et al.:
RNAs coordinate nuclear envelope assembly and DNA replication through ELYS recruitment to chromatin.
Nat Commun.
2017; 8(1): 2130. PubMed Abstract
| Publisher Full Text
| Free Full Text
86.
Sansam CG, Goins D, Siefert JC, et al.:
Cyclin-dependent kinase regulates the length of S phase through TICRR/TRESLIN phosphorylation.
Genes Dev.
2015; 29(5): 555–66. PubMed Abstract
| Publisher Full Text
| Free Full Text
91.
Mac Auley A, Werb Z, Mirkes PE:
Characterization of the unusually rapid cell cycles during rat gastrulation.
Development.
1993; 117(3): 873–83. PubMed Abstract
93.
Dungrawala H, Rose KL, Bhat KP, et al.:
The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability.
Mol Cell.
2015; 59(6): 998–1010. PubMed Abstract
| Publisher Full Text
| Free Full Text
94.
Aranda S, Rutishauser D, Ernfors P:
Identification of a large protein network involved in epigenetic transmission in replicating DNA of embryonic stem cells.
Nucleic Acids Res.
2014; 42(11): 6972–86. PubMed Abstract
| Publisher Full Text
| Free Full Text
97.
Kermi C, Prieto S, van der Laan S, et al.:
RAD18 Is a Maternal Limiting Factor Silencing the UV-Dependent DNA Damage Checkpoint in Xenopus Embryos.
Dev Cell.
2015; 34(3): 364–72. PubMed Abstract
| Publisher Full Text
Research in DM’s laboratory is supported by grants from “Fondation ARC pour la Recherche sur le Cancer”, La Ligue contre le Cancer, INSERM, and MSD Avenir.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Aze A and Maiorano D. Recent advances in understanding DNA replication: cell type–specific adaptation of the DNA replication program [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1351 (https://doi.org/10.12688/f1000research.15408.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
MacAlpine DM and Hoffman R. Reviewer Report For: Recent advances in understanding DNA replication: cell type–specific adaptation of the DNA replication program [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1351 (https://doi.org/10.5256/f1000research.16790.r37391)
We confirm that we have read this submission and believe that we have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
We confirm that we have read this submission and believe that we have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
MacAlpine DM and Hoffman R. Reviewer Report For: Recent advances in understanding DNA replication: cell type–specific adaptation of the DNA replication program [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1351 (https://doi.org/10.5256/f1000research.16790.r37391)
Speck C. Reviewer Report For: Recent advances in understanding DNA replication: cell type–specific adaptation of the DNA replication program [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1351 (https://doi.org/10.5256/f1000research.16790.r37392)
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Speck C. Reviewer Report For: Recent advances in understanding DNA replication: cell type–specific adaptation of the DNA replication program [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1351 (https://doi.org/10.5256/f1000research.16790.r37392)
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)