Keywords
Cryptosporidium, cell culture, genetic modification, transcriptomics, drug discovery
Cryptosporidium, cell culture, genetic modification, transcriptomics, drug discovery
The intestinal apicomplexan parasite Cryptosporidium is responsible for waterborne outbreaks of diarrheal disease worldwide and continues to cause opportunistic infection in immunocompromised hosts, including patients with untreated HIV/AIDS1. Recently, this parasite has been increasingly recognized as a major cause of diarrhea with long-term consequences, such as malnutrition, growth, and cognitive deficits in young children in resource-limited settings2–7. Despite the global burden of cryptosporidiosis, treatment options are limited to supportive therapy and a single US Food and Drug Administration-approved drug, nitazoxanide, which has limited efficacy in malnourished children and is ineffective in immunocompromised individuals8–11. Thus, there is an urgent need for the development of novel strategies to control cryptosporidiosis, particularly in susceptible populations4. Progress in this field has been severely hampered by the notorious intractability of the parasite and limited tools to study it4. Recently, however, there have been some exciting technological breakthroughs which have spurred renewed efforts to understand Cryptosporidium at the molecular and cellular levels and the pathogenesis of cryptosporidiosis and to develop new approaches to combat this disease. This article reviews these breakthroughs and discusses the continuing challenges associated with Cryptosporidium research.
There are several constraints to drug development for cryptosporidiosis4,12. The pathogenesis of the disease and the molecules and pathways that can be targeted for drug development are poorly understood13. One of the reasons for this is the lack of primary intestinal epithelial cell (IEC) models that recapitulate normal human IEC structure and function and support robust infection and the completion of the life cycle of the parasite, permitting continuous propagation in vitro. Most models of C. parvum infection in vitro employ transformed or immortalized adenocarcinoma-derived human IEC lines, such as Caco-2, HCT-8, and HT2914,15. Primary human and bovine IEC models permit infections and Cryptosporidium hominis and C. parvum, respectively16,17, and C. parvum infection in a non-cancer-derived IEC line, FHs 74 Int, has also been reported18. However, these cell lines support C. parvum infection for only a few days, do not permit completion of the life cycle or continuous propagation12, and can display variation in gene expression depending on culture conditions19. Cell-free systems for C. parvum culture have been reported20, but although the parasite was observed to complete its life cycle by transmission electron microscopy21, these in vitro axenic systems do not permit investigations of the parasite interactions with the host epithelia.
Recently, however, promising cell culture models that overcome some of the limitations of earlier models have been developed. For instance, Morada et al.22 described a culture system in a simulated gut-like environment using HCT-8 cells and polysulfone hollow-fiber technology for continuous and long-term production of oocysts. Through the use of specialized equipment and culture medium supplements, C. parvum oocysts could be continuously propagated for more than 6 months. Various developmental stages were identified by scanning electron microscopy of infected host cells after 8 weeks of parasitic growth and by antibody staining using anti-sporozoite and anti-oocyst wall antibodies SPORO-GLO and CRYPT-A-GLO (http://waterborneinc.com/crypt-a-glo/), respectively. This system promises to be useful for large-scale generation of infective oocysts. Other potential advantages include the availability of a large surface area for efficient growth of host cells at high cell densities comparable to in vivo conditions and the creation of a biphasic medium that mimics the anaerobic gut environment. However, this technology is not readily scalable and is not practical for the screening of drugs or for investigations of pathogenesis in multiple replicates in real time. In addition, the hollow-fiber system employs the transformed HCT-8 cell line that may not replicate the structure and function of primary human intestinal cells.
Miller et al.23 developed a culture system for the propagation of infective C. parvum oocysts employing the human esophageal squamous cell carcinoma cell line COLO-680N. Infection with two different C. parvum strains resulted in the production of sufficient amounts of infective oocysts (identified by CRYPT-A-GLO) to enable continuous propagation of the parasite. Lipidomics and atomic force microscopy imaging confirmed the presence of oocysts in the culture. This seems to be a promising model for propagation of oocysts, but it uses an esophageal squamous carcinoma-derived line rather than primary human IECs, the natural habitat of anthroponotic Cryptosporidium spp.
Two-dimensional culture systems are not ideal, since they do not represent the three-dimensional (3D) topology of the intestine in vivo. DeCicco Repass et al.24 used a novel 3D bioengineered human intestinal tissue system for C. parvum infection. This model employs a porous silk protein scaffolding system with a lumen that is seeded with human IEC lines Caco-2/HT29-MTX and a “bulk” space surrounding the lumen that is seeded with human myofibroblasts (H-InMyoFibs) which secrete growth factors that support IEC growth25 (Figure 1). Immunofluorescence staining using monoclonal antibody (mAb) 4E9 which recognizes a glycopeptide epitope26 and confocal microscopy was used to identify invasive and intracellular C. parvum stages. Infection in this model lasted for at least 17 days (the longest time tested). Importantly, contents from infected scaffolds could be transferred to fresh scaffolds to establish new infections for at least three rounds, suggesting that infection might be propagated for a longer term. Completion of the life cycle, with the formation of new oocysts identified by CRYPT-A-GLO, occurred in this model, which can be used to evaluate pathogenic processes and appears to be amenable to rapid drug screening. However, the small size of the model is a limitation for large-scale propagation. Additionally, the culture system employs transformed adenocarcinoma-derived Caco-2 and HT29-MTX cells, which are not representative of primary human IEC cells in vivo12.
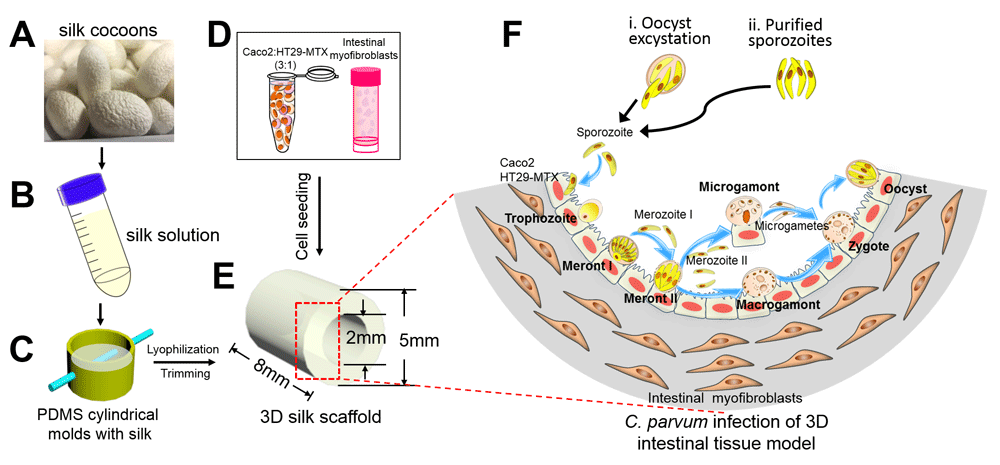
Silk cocoons (A) were processed to yield a viscous silk solution (B). (C) The silk solution was poured into cylindrical molds, and a wire was inserted to develop a lumen equivalent. Caco-2 and HT29-MTX cells (D) were seeded into the lumen (E), while the porous bulk space was seeded with H-InMyoFibs. (F) The Caco-2 and HT29-MTX cells in the lumen were infected with Cryptosporidium parvum oocysts or purified sporozoites. Intracellular development through asexual and sexual cycles occurred to complete the life cycle with the formation of oocysts. 3D, three-dimensional; PDMS, polydimethylsiloxane. Figure reproduced with permission from the American Society for Microbiology23.
This issue was addressed using primary human intestinal enteroid cells to replace the transformed IEC lines in the system27. Enteroids are stem cell-derived 3D structures that can be generated from crypts derived from human intestinal biopsies, can be passaged indefinitely, and are a more physiological alternative to transformed cell lines28,29. This 3D ex vivo model supports robust C. parvum infection and results in the production of oocysts30. The use of murine-derived enteroids for C. parvum infection has also been reported31. Recently, Heo et al. used human lung and intestinal epithelial organoids and CRYPT-A-GLO and SPRO-GLO antibodies to demonstrate that these systems can support the complete life cycle of C. parvum32. Although the newly generated oocysts were infectious in vivo, the yield and efficiency of infection were low, and compared with the hollow-fiber system, these organoids could support continuous culture for only 28 days concurrent with a decrease in parasite number over time.
Until recently, reverse genetics approaches were not tractable for Cryptosporidium, making the elucidation of virulence factors and pathogenic pathways and the validation of drug targets next to impossible4,13. In a transformative breakthrough for the field (Figure 2), Vinayak et al.33 tested their optimized transfection protocol by targeting the thymidine kinase (TK) gene of C. parvum. Parasites were transfected with a vector encoding a guide RNA specific to TK and the Streptococcus pyogenes Cas9 endonuclease, which introduces a double-stranded break into the TK locus. Expression of the guide RNA and of Cas9 was driven by C. parvum promoters. In addition, parasites were co-transfected with a donor DNA, which serves as a template to repair the double-stranded break introduced by Cas9. This donor DNA encoded a fusion protein of the Nluc luciferase reporter gene and the NeoR resistance gene, whose expression was driven by the C. parvum enolase promoter. To promote homologous recombination, the donor DNA included flanking sequences complementary to regions upstream and downstream of the endogenous TK gene. This work resulted in the successful creation of a stable C. parvum TK knockout line33.
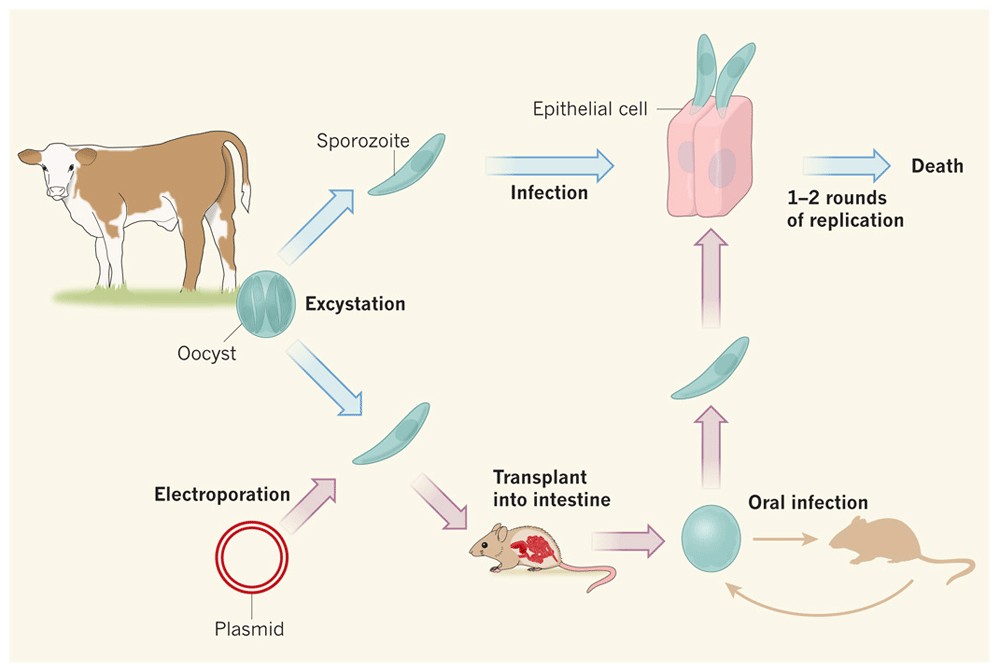
Cryptosporidium parvum oocysts isolated from the feces of infected calves can be excysted to release sporozoites which can infect mammalian epithelial cells in culture but only for one or two rounds of replication before they die. Vinayak et al. used CRISPR/Cas9 technology to genetically modify C. parvum sporozoites33. Selection and replication of modified parasites requires direct injection into surgically isolated intestines of interferon-gamma knockout mice. Modified oocysts collected from mouse feces can be analyzed in culture or used to inoculate new mice to maintain the transgenic line. Figure reproduced with permission from Springer39.
Several components of the canonical RNA interference (RNAi) pathway are not encoded in Cryptosporidium genomes, precluding RNAi mechanisms for gene silencing34,35. In this pathway, double-stranded RNA (dsRNA) with sequence complementarity to an mRNA of interest is processed into single-stranded RNA (ssRNA), leading to the cleavage of the target mRNA by the enzyme Argonaute 2 (Ago2). Castellanos-Gonzalez et al.36 addressed this limitation by transfecting parasites with human Ago2 protein, which was preloaded with ssRNAs against C. parvum transcripts, resulting in the reduced expression of targeted transcripts.
Another molecular mechanism of gene silencing was developed for C. parvum through the use of morpholinos37,38. Morpholinos are synthetic DNA analogs that inhibit protein translation initiation by base pairing to complementary mRNA. In a recent study, excysted sporozoites and HCT-8 cells were treated with morpholinos targeting C. parvum lactate dehydrogenase (CpLDH) and putative arginine n-methyltransferase (CpAMT) prior to in vitro infection, resulting in decreased protein expression of CpLDH and CpAMT37. In a follow-up study, morpholinos optimized for in vivo delivery against CpLDH and Cp15/60 were injected intraperitoneally into interferon-gamma knockout mice, which then were infected with C. parvum oocysts. Expression of both proteins decreased following infection in morpholino-treated mice38. Whether or not protein expression rebounds during the course of subsequent in vitro infection using these purified oocysts was not ascertained.
To elucidate parasite pathways that are important for pathogenesis, it is essential to examine the gene expression profile of C. parvum during infection. RNA sequencing (RNA-Seq) has been an invaluable high-throughput, deep-sequencing-based approach to mapping the full cellular transcriptome. This big data approach allows researchers to probe host–pathogen interactions by analyzing transcriptomic changes in both host and pathogen at any point in time. In a recent study40, RNA from C. parvum infection of the porcine intestinal cell line IPEC-J2 was harvested 24 hours after infection and subjected to RNA-Seq analysis. The transcriptomes of oocysts alone and uninfected IPEC-J2 cells were also mapped as controls. This study identified genes involved in ribosome biogenesis and translation as being upregulated in C. parvum during infection in vitro compared with oocysts. In addition, cell division pathways were upregulated in infected versus uninfected IPEC-J2 cells40. C. parvum transcripts accounted for only 2.2% of the total reads in this study, but whether that proportion was sufficient to measure low-level transcripts was not addressed. Another RNA-Seq study compared the transcriptome of C. parvum sporozoites purified from oocysts, the intestine of infected calves, or infected HCT-8 cells41. General metabolic pathways were upregulated during in vivo infection compared with sporozoites alone. In addition, mucins, which have been shown to be involved in host cell attachment13, were upregulated in vivo. Genes encoding oocyst wall proteins were upregulated during both in vitro and in vivo infection compared with sporozoites41. However, this study did not assess transcriptomic changes in host cells.
Development of CRISPR/Cas9-based technology for genetic modification of C. parvum has paved the way for evaluating promising drug candidates using transgenic parasites in animal models42 (Figure 3). In a recent report, KDU731, a pyrazolopyridine, inhibited the enzymatic activity of recombinant C. parvum phosphatidylinositol 4-kinase (PI4K), exhibited anti-cryptosporidial activity in vitro, and was pharmacologically non-toxic43. Treatment with KDU731 of interferon-gamma knockout mice infected with transgenic C. parvum resulted in a marked decrease in intestinal colonization and diminished oocyst shedding43. Moreover, KDU731 was efficacious in a calf model of cryptosporidiosis, leading to decreased oocyst shedding and a reduction in diarrhea. However, treatment did not entirely eliminate parasite shedding or diarrhea in these animals43.

(1) A high content imaging infection assay in vitro was used to identify Cryptosporidium parvum inhibitory compounds. (2) Secondary screening using a cytopathic effect-based assay was used to identify imidazopyrazines and pyrazolopyridines with inhibitory activity against C. parvum and Cryptosporidium hominis. (3) Identification, expression, and enzymatic activity of the C. parvum phosphatidylinositol-4-OH kinase (PI4K). (4) Pharmacokinetics and toxicity testing of KDU731. (5) Activity of KDU731 against transgenic C. parvum infection in interferon-gamma knockout mice. (6) Activity of KDU731 against native C. parvum infection in the neonatal calf model. (7) Additional preclinical evaluation is needed before initiation of human clinical trials. Figure reproduced with permission from Elsevier42.
Bumped kinase inhibitors (BKIs) are a series of compounds that inhibit calcium-dependent protein kinase 1 (CDPK1) activity in apicomplexan parasites. Several studies to determine the efficacy and safety of BKI in the treatment of cryptosporidiosis have been published recently44–47. Several of these compounds have been found to reduce the severity of diarrhea and fecal oocyst shedding in calf and piglet models of cryptosporidiosis caused by C. parvum and C. hominis44,45,47. Unfortunately, the compounds tested were not fully curative, and many displayed cytotoxicity, cardiotoxicity, or fetal toxicity (or a combination of these) in a variety of safety tests44,45,47.
Recently, MMV665917, from the Medicines for Malaria Venture “Malaria Box” of drugs48, was found to be efficacious against cryptosporidiosis in immunocompromised mice and neonatal calf models49,50. The severity of diarrhea and oocyst shedding in the calf model was reduced but not eliminated50. The mechanism of action of MMV665917 remains unknown, and toxicity assessments must be performed to establish safety in humans.
An alternative approach to drug discovery is to screen for inhibition of a specific parasite target. A screen of the Prestwick Chemical Library for compounds that inhibit recombinant C. parvum glucose-6-phosphate isomerase (GPI) activity identified ebselen, a selenium-containing aromatic compound, which inhibited C. parvum, but not human, recombinant GPI activity51. Likewise, ebselen inhibited C. parvum growth during in vitro infection51. However, the selectivity index—the ratio of 50% HCT-8 toxicity to 50% parasite inhibition—was only about fourfold, and efficacy in animal models was not ascertained51.
Despite these recent advances, a number of limitations persist and should be addressed in future studies. For example, none of the newer cell culture systems is ideal in all respects. Many of these systems have limited visualization of parasite growth and development. Most models employ antibody-based detection by immunofluorescence. Until recently, specific antibodies to most developmental stages were not available. Wilke et al. recently reported development of C. parvum stage-specific murine mAbs52 which will be useful for identifying the intracellular stages present in the various models. Similarly, scanning or transmission electron microscopy relies on morphological assessment of which stages can be visualized. With the current availability of genetic manipulation technology for Cryptosporidium, epitope tagging with fluorescent tags or tags which can be fluorescently labeled with specific antibodies can be used to assess the presence of various stages.
The 3D bioengineered model, employing primary human enteroid cells, is one of the most exciting approaches but could be enhanced by incorporating additional cell types (such as nerve, immune, and endothelial cells), co-culturing with gut microbiota, and introducing physiological conditions (such as flow, peristalsis, and low oxygen tension) to develop an integrated “mini-gut” system.
Generating transgenic C. parvum strains using CRISPR/Cas9-based technology will likely advance and transform research on this important parasite33,39,53. However, the selection and maintenance of transgenic strains are currently laborious and expensive. Transgenic C. parvum strains cannot be propagated continually in vitro, requiring direct injection of transfected sporozoites into the surgically exposed small intestine of paromomycin-treated, interferon-gamma knockout mice for selection and propagation33. Importantly, if a target gene is essential for the invasion of small intestinal cells, it is currently not possible to generate stable transgenic strains of these parasites. Strategies for generating conditional knockouts or for complementing deleted genes have not yet been reported. NeoR, conferring resistance against paromomycin, is the only selectable marker currently available. Of the newer techniques for genetic manipulation of C. parvum, only the CRISPR/Cas9-based gene deletion has been reproduced in another system43, and this remains the only technique for targeted deletion, as opposed to knockdown of C. parvum genes. Finally, genetic manipulation of C. hominis, the species which is most relevant for human infection, is not yet possible.
For RNA-Seq analysis to be of optimal value, a high enough proportion of host cells must be infected in order for transcriptomic changes in the host to be detected. Likewise, in order to accurately quantify changes in the transcriptome of the parasite, the proportion of parasite transcripts to host transcripts must constitute a threshold of the total RNA pool to be analyzed. These issues might be overcome by applying single-cell RNA-Seq and increasing sequencing depth (or enriching for parasite transcripts prior to sequencing or both). In such an approach, RNA-Seq-based investigations should allow scientists to explore unknown genes or pathways that are essential for survival of the parasite and thus represent potential drug targets.
Significant advances have been made in the search for novel drugs for the treatment of cryptosporidiosis. However, some of these candidates do not eliminate oocyst shedding, lead to only modest reduction in diarrhea severity or duration (or both), or have been neither rigorously tested for toxicity nor evaluated in pre-clinical trials. Nonetheless, many of these compounds are modifiable, and it might be possible to develop new analogs with enhanced efficacy and decreased toxicity to human cells. To the best of our knowledge, none of the drugs has reached the human clinical trial stage. In an effort to identify additional novel targets or inhibitors (or both), further studies using transgenic, transcriptomic, and drug screening tools are warranted.
Here, we have reviewed recent, promising advances in novel cell culture systems, genetic and molecular techniques, and drug discovery for Cryptosporidium. Space constraints preclude an appraisal of other recent advances in the clinical, diagnostic, and epidemiological aspects of cryptosporidiosis, the biochemistry and cellular biology of the parasite, and the innate and adaptive immune responses to infection. However, the present review shows that recent innovations are advancing knowledge of Cryptosporidium and provide a basis for the development of effective and practical strategies for the prevention and control of cryptosporidiosis in the vulnerable populations who need them most.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)