Keywords
DNA methylation, differential methylation, epigenetics, causal inference, variable importance, machine learning, targeted loss-based estimation
This article is included in the Bioconductor gateway.
This article is included in the RPackage gateway.
DNA methylation, differential methylation, epigenetics, causal inference, variable importance, machine learning, targeted loss-based estimation
DNA methylation is a fundamental epigenetic process known to play an important role in the regulation of gene expression. DNA methylation most commonly occurs at CpG sites and involves the addition of a methyl group (CH3) to the fifth carbon of the cytosine ring structure to form 5-methylcytosine. Numerous biological and medical studies have implicated DNA methylation as playing a role in disease and development1. Perhaps unsurprisingly then, biotechnologies have been developed to rigorously probe the molecular mechanisms of this epigenetic process. Modern assays, like the Illumina Infinium HumanMethylation BeadChip assay, allow for quantitative interrogation of DNA methylation, at single-nucleotide resolution, across a comprehensive set of CpG sites scattered across the genome; moreover, the computational biology community has invested significant effort in the development of tools for properly removing technological effects that may contaminate biological signatures measured by such assays [2, Dedeurwaerder et al.3]. Despite these advances in both biological and bioninformatical techniques, most statistical methods available for differential analysis of data produced by such assays rely on over-simplified models that do not readily extend to such high-dimensional data structures without restrictive modeling assumptions and the use of inferentially costly hypothesis testing corrections. When these standard assumptions are violated, estimates of the population-level effect of an exposure or treatment may suffer from large bias. What’s more, reliance on restrictive and misspecified statistical models naturally leads to biased effect estimates that are not only misleading in assessing effect sizes but also result in false discoveries as these biased estimates are subject to testing and inferential procedures. Such predictably unreliable methods serve only to produce findings that are later invalidated by replication studies and add still further complexity to discovering biological targets for potential therapeutics. Data-adaptive estimation procedures that utilize machine learning provide a way to overcome many of the problems common in classical methods, controlling for potential confounding even in high-dimensional settings; however, interpretable statistical inference (i.e., confidence intervals and hypothesis tests) from such data-adaptive estimates is challenging to obtain4.
In this paper, we briefly present an alternative to such statistical analysis approaches in the form of a nonparametric estimation procedure that provides simple and readily interpretable statistical inference, discussing at length a recent implementation of the methodology in the methyvim R package. Inspired by recent advances in statistical causal inference and machine learning, we provide a computationally efficient technique for obtaining targeted estimates of nonparametric variable importance measures (VIMs)5, estimated at a set of pre-screened CpG sites, controlling for the False Discovery Rate (FDR) as if all sites were tested. Under standard assumptions (e.g., identifiability, strong ignorability)6, targeted minimum loss-based estimators of regular asymptotically linear estimators have sampling distributions that are asymptotically normal, allowing for reliable point estimation and the construction of Wald-style confidence intervals [7, van der Laan and Rose8]. In the context of DNA methylation studies, we define the counterfactual outcomes under a binary treatment as the observed methylation (whether Beta- or M-) values a CpG site would have if all subjects were administered the treatment and the methylation values a CpG site would have if treatment were withheld from all subjects. Although these counterfactual outcomes are, of course, impossible to observe, they do have statistical analogs that may be reliably estimated (i.e., identified) from observed data under a small number of untestable assumptions6. We describe an algorithm that incorporates, in its final step, the use targeted minimum loss-based estimators (TMLE)9 of a given VIM of interest, though we defer rigorous and detailed descriptions of this aspect of the statistical methodology to work outside the scope of the present manuscript [9, van der Laan and Rose7, van der Laan and Rose8]. The proposed methodology assesses the individual importance of a given CpG site, as a proposed measure of differential methylation, by utilizing state-of-the-art machine learning algorithms in deriving targeted estimates and robust inference of a VIM, as considered more broadly for biomarkers in Bembom et al.10 and Tuglus and van der Laan11. In the present work, we focus on the methyvim software package, available through the Bioconductor project [12, Huber et al.13] for the R language and environment for statistical computing14, which implements a particular realization of this methodology specifically tailored for the analysis and identification of differentially methylated positions (DMPs).
For an extended discussion of the general framework of targeted minimum loss-based estimation and detailed accounts of how this approach may be brought to bear in developing answers to complex scientific problems through statistical and causal inference, the interested reader is invited to consult van der Laan and Rose7 and van der Laan and Rose8. For a more general introduction to causal inference, Pearl6 and Hernan and Robins15 may be of interest.
The core functionality of this package is made available via the eponymous methyvim function, which implements a statistical algorithm designed to compute targeted estimates of VIMs, defined in such a way that the VIMs represent parameters of scientific interest in computational biology experiments; moreover, these VIMs are defined such that they may be estimated in a manner that is very nearly assumption-free, that is, within a fully nonparametric statistical model. The statistical algorithm consists of several major steps summarized below. Additional methodological details on the use of targeted minimum loss-based estimation in this problem setting is provided in Supplementary File 1.
1. Pre-screening of genomic sites is used to isolate a subset of sites for which there is cursory evidence of differential methylation. Currently, the available screening approach adapts core routines from the limma R package. Following the style of the function for performing screening via limma, users may write their own screening functions and are invited to contribute such functions to the core software package by opening pull requests at the GitHub repository: https://github.com/nhejazi/methyvim.
2. Nonparametric estimates of VIMs, for the specified target parameter, are computed at each of the CpG sites passing the screening step. The VIMs are defined in such a way that the estimated effects is of an binary treatment on the methylation status of a target CpG site, controlling for the observed methylation status of the neighbors of that site. Currently, routines are adapted from the tmle R package.
3. Since pre-screening is performed prior to estimating VIMs, we apply the modified marginal Benjamini and Hochberg step-up False Discovery Rate controlling procedure for multi-stage analyses (FDR-MSA), which is well-suited for avoiding false positive discoveries when testing is only performed on a subset of potential targets.
Parameters of Interest For CpG sites that pass the pre-screening step, a user-specified target parameter of interest is estimated independently at each site. In all cases, an estimator of the parameter of interest is constructed via targeted minimum loss-based estimation.
Two popular target causal parameters for discrete-valued treatments or exposures are
The average treatment effect (ATE): The effect of a binary exposure or treatment on the observed methylation at a target CpG site is estimated, controlling for the observed methylation at all other CpG sites in the same neighborhood as the target site, based on an additive form. Often denoted , the parameter estimate represents the additive difference in methylation that would have been observed at the target site had all observations received the treatment versus the counterfactual under which none received the treatment.
The relative risk (RR): The effect of a binary exposure or treatment on the observed methylation at a target CpG site is estimated, controlling for the observed methylation at all other CpG sites in the same neighborhood as the target site, based on a geometric form. Often denoted, , the parameter estimate represents the multiplicative difference in methylation that would have been observed at the target site had all observations received the treatment versus the counterfactual under which none received the treatment.
Estimating the VIM corresponding to the parameters above, for discrete-valued treatments or exposures, requires two separate regression steps: one for the treatment mechanism (propensity score) and one for the outcome regression. Technical details on the nature of these regressions are discussed in Hernan and Robins15, and details for estimating these regressions in the framework of targeted minimum loss-based estimation are discussed in van der Laan and Rose7.
Class methytmle We have adopted a class methytmle to help organize the functionality within this package. The methytmle class builds upon the GenomicRatioSet class provided by the minfi package so all of the slots of GenomicRatioSet are contained in a methytmle object. The new class introduced in the methyvim package includes several new slots:
call - the form of the original call to the methyvim function.
screen_ind - indices identifying CpG sites that pass the screening process.
clusters - non-unique IDs corresponding to the manner in wich sites are treated as neighbors. These are assigned by genomic distance (bp) and respect chromosome boundaries (produced via a call to bumphunter::clusterMaker).
var_int - the treatment/exposure status for each subject. Currently, these must be binary, due to the definition of the supported targeted parameters.
param - the name of the target parameter from which the estimated VIMs are defined.
vim - a table of statistical results obtained from estimating VIMs for each of the CpG sites that pass the screening procedure.
ic - the measured array values for each of the CpG sites passing the screening, transformed into influence curve space based on the chosen target parameter.
The show method of the methytmle class summarizes a selection of the above information for the user while masking some of the wealth of information given when calling the same method for GenomicRatioSet. All information contained in GenomicRatioSet objects is preserved in methytmle objects, so as to ease interoperability with other differential methylation software for experienced users. We refer the reader to the package vignette, “methyvim: Targeted Data-Adaptive Estimation and Inference for Differential Methylation Analysis,” included in any distribution of the software package, for further details.
To examine the practical applications and the full set of utilities of the methyvim package, we will use a publicly available example data set produced by the Illumina 450K array, from the minfiData R package, accessible via the Bioconductor project at https://doi.org/doi:10.18129/B9.bioc.minfiData.
Preliminaries: Setting up the data We begin by loading the package and the data set. After loading the data, which comes in the form of a raw MethylSet object, we perform some further processing by mapping to the genome (with mapToGenome) and converting the values from the methylated and unmethylated channels to Beta-values (via ratioConvert). These two steps together produce an object of class GenomicRatioSet, provided by the minfi package.
suppressMessages( # numerous messages displayed at time of loading library(minfiData) ) data(MsetEx) mset <- mapToGenome(MsetEx) grs <- ratioConvert(mset) grs
## class: GenomicRatioSet
## dim: 485512 6
## metadata(0):
## assays(2): Beta CN
## rownames(485512): cg13869341 cg14008030 ... cg08265308 cg14273923
## rowData names(0):
## colnames(6): 5723646052_R02C02 5723646052_R04C01 ...
## 5723646053_R05C02 5723646053_R06C02
## colData names(13): Sample_Name Sample_Well ... Basename filenames
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19
## Preprocessing
## Method: Raw (no normalization or bg correction)
## minfi version: 1.21.2
## Manifest version: 0.4.0We can create an object of class methytmle from any GenomicRatioSet object simply invoking the S4 class constructor .methytmle:
library(methyvim)
## methyvim v1.3.1: Targeted, Robust, and Model-free Differential Methylation Analysisgrs_mtmle <- .methytmle(grs) grs_mtmle
## class: methytmle
## dim: 485512 6
## metadata(0):
## assays(2): Beta CN
## rownames(485512): cg13869341 cg14008030 ... cg08265308 cg14273923
## rowData names(0):
## colnames(6): 5723646052_R02C02 5723646052_R04C01 ...
## 5723646053_R05C02 5723646053_R06C02
## colData names(13): Sample_Name Sample_Well ... Basename filenames
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19
## Preprocessing
## Method: Raw (no normalization or bg correction)
## minfi version: 1.21.2
## Manifest version: 0.4.0
## Target Parameter:
## Results:
## Object of class "data.frame"
## data frame with 0 columns and 0 rowsAdditionally, a GenomicRatioSet can be created from a matrix with the function makeGenomicRatioSetFromMatrix provided by the minfi package.
Differential Methylation Analysis For this example analysis, we’ll treat the condition of the patients as the exposure/treatment variable of interest. The methyvim function requires that this variable either be numeric or easily coercible to numeric. To facilitate this, we’ll simply convert the covariate (currently a character):
var_int <- (as.numeric(as.factor(colData(grs)$status)) - 1)
n.b., the re-coding process results in “normal” patients being assigned a value of 1 and cancer patients a 0.
Now, we are ready to analyze the effects of cancer status on DNA methylation using this data set. We proceed as follows with a targeted minimum loss-based estimate of the Average Treatment Effect.
methyvim_cancer_ate <- methyvim(data_grs = grs, var_int = var_int, vim = "ate", type = "Beta", filter = "limma", filter_cutoff = 0.20, obs_per_covar = 2, parallel = FALSE, sites_comp = 250, tmle_type = "glm" )
## Loading required package: nnlsNote that we set the obs_per_covar argument to a relatively low value (just 2, even though the recommended value, and default, is 20) for the purposes of this example as the sample size is only 10. We do this only to exemplify the estimation procedure and it is important to point out that such low values for obs_per_covar will compromise the quality of inference obtained because this setting directly affects the definition of the target parameter.
Further, note that here we apply the glm flavor of the tmle_type argument, which produces faster results by fitting models for the propensity score and outcome regressions using a limited number of parametric models. By contrast, the sl (for “Super Learning”) flavor fits these two regressions using highly nonparametric and data-adaptive procedures (i.e., via machine learning). Obtaining the estimates via GLMs results in each of the regression steps being less robust than if nonparametric regressions were used.
We can view a table of results by examining the vim slot of the produced object, most easily displayed by simply printing the resultant object:
methyvim_cancer_ate## class: methytmle
## dim: 485512 6
## metadata(0):
## assays(2): Beta CN
## rownames(485512): cg13869341 cg14008030 ... cg08265308 cg14273923
## rowData names(0):
## colnames(6): 5723646052_R02C02 5723646052_R04C01 ...
## 5723646053_R05C02 5723646053_R06C02
## colData names(13): Sample_Name Sample_Well ... Basename filenames
## Annotation
## array: IlluminaHumanMethylation450k
## annotation: ilmn12.hg19
## Preprocessing
## Method: Raw (no normalization or bg correction)
## minfi version: 1.21.2
## Manifest version: 0.4.0
## Target Parameter: Average Treatment Effect
## Results:
## lwr_ci est_ate upr_ci var_ate
## cg14008030 -1.195660e-01 -0.0314159619 0.0567340489 2.022705e-03
## cg20253340 -8.850637e-02 -0.0588661418 -0.0292259165 2.286919e-04
## cg21870274 -9.499057e-02 -0.0291189817 0.0367526091 1.129495e-03
## cg17308840 -4.626018e-02 -0.0071524518 0.0319552773 3.981191e-04
## cg00645010 -2.677328e-02 -0.0134655543 -0.0001578256 4.609945e-05
## cg27534567 6.745648e-02 0.1157118536 0.1639672225 6.061486e-04
## cg08258224 1.365045e-01 0.3050884951 0.4736724770 7.398105e-03
## cg20275697 -3.021026e-01 -0.1231299608 0.0558427144 8.337989e-03
## cg24373735 -4.283533e-02 0.0076666975 0.0581687266 6.639043e-04
## cg12445832 -6.082411e-02 0.0150395574 0.0909032203 1.498151e-03
## cg01097950 -4.392159e-02 0.0658323796 0.1755863507 3.135656e-03
## cg01782097 -1.082072e-02 0.0010232901 0.0128672954 3.651615e-05
## pval n_neighbors n_neighbors_control max_cor_neighbors
## cg14008030 4.848468e-01 0 0 NA
## cg20253340 9.917426e-05 0 0 NA
## cg21870274 3.862537e-01 2 1 0.94435796
## cg17308840 7.199943e-01 2 1 0.94435796
## cg00645010 4.734007e-02 2 2 0.52368097
## cg27534567 2.602936e-06 1 0 0.93629683
## cg08258224 3.895914e-04 1 0 0.93629683
## cg20275697 1.775155e-01 0 0 NA
## cg24373735 7.660489e-01 0 0 NA
## cg12445832 6.976022e-01 0 0 NA
## cg01097950 2.397377e-01 0 0 NA
## cg01782097 8.655302e-01 1 1 -0.39410834
## [ reached getOption("max.print") -- omitted 238 rows ]Finally, we may compute FDR-corrected p-values, by applying a modified procedure for controlling the False Discovery Rate for multi-stage analyses (FDR-MSA)16. We do this by simply applying the fdr_msa function.
fdr_p <- fdr_msa(pvals = vim(methyvim_cancer_ate)$pval, total_obs = nrow(methyvim_cancer_ate))
Having explored the results of our analysis numerically, we now proceed to use the visualization tools provided with the methyvim R package to further enhance our understanding of the results.
Visualization of results While making allowance for users to explore the full set of results produced by the estimation procedure (by way of exposing these directly to the user), the methyvim package also provides three (3) visualization utilities that produce plots commonly used in examining the results of differential methylation analyses.
A simple call to plot produces side-by-side histograms of the raw p-values computed as part of the estimation process and the corrected p-values obtained from using the FDR-MSA procedure.
plot(methyvim_cancer_ate, type = "raw_pvals")
plot(methyvim_cancer_ate, type = "fdr_pvals")
Remark: The plots displayed above may also be generated as side-by-side histograms in a single plot object. This is the default for the plot method and may easily be invoked by specifying no additional arguments to the plot function, unlike in the above.
From the code snippets displayed above, Figure 1 displays a histogram of raw (or uncorrected) p-values from hypothesis testing of the statistical parameter corresponding to the average treatment effect while Figure 2 produces a histogram of p-values from the same set of hypothesis tests, correcting for multiple testing using the FDR-MSA method16. While histograms of the p-values may be generally useful in inspecting the results of the estimation procedure, a more common plot used in examining the results of differential methylation procedures is the volcano plot, which plots the parameter estimate along the x-axis and −log10(p-value) along the y-axis. We implement such a plot in the methyvolc function:
methyvolc(methyvim_cancer_ate)
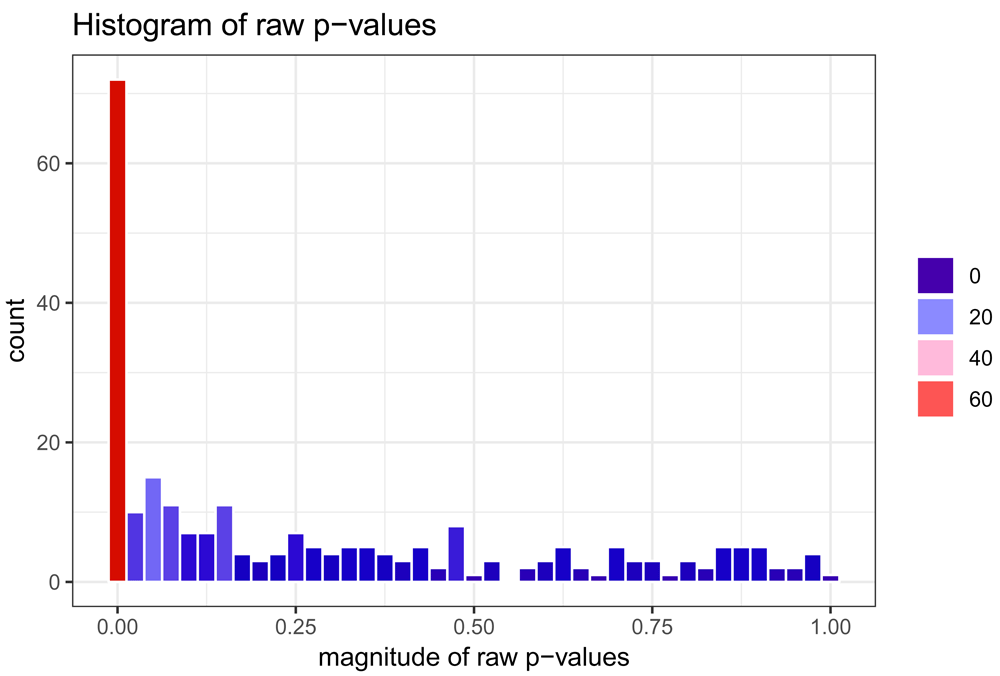

Figure 3 above displays a volcano plot of the raw (or unadjusted) p-values against estimates of the effect of interest (by default, the average treatment effect in methyvim). The purpose of such a plot is to ensure that very low (possibly statistically significant) p-values do not arise from cases of low variance. This appears to be the case in the plot above (notice that most parameter estimates are near zero, even in cases where the raw p-values are quite low).

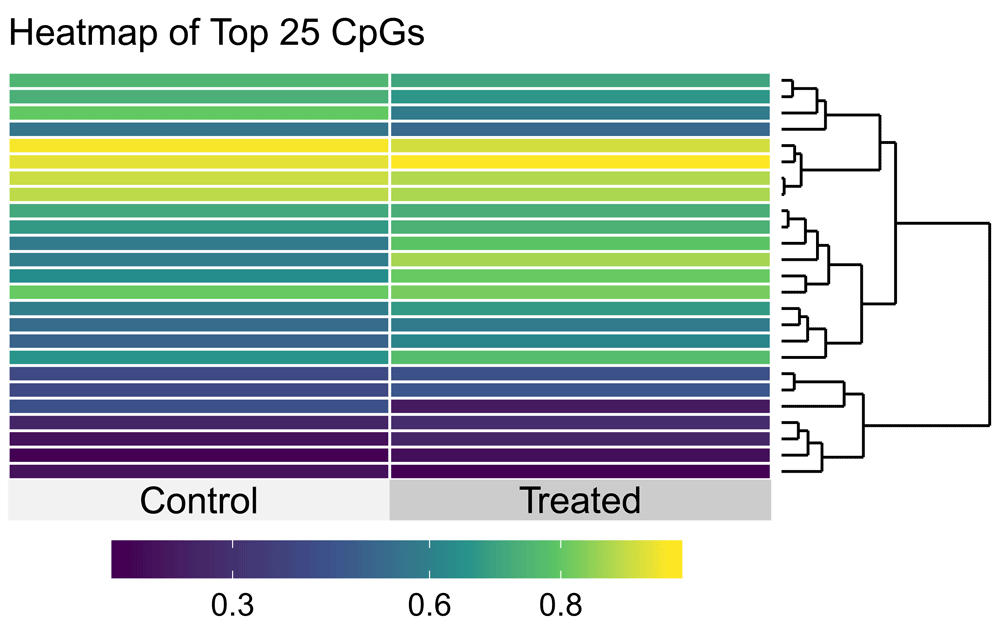
Yet another popular plot for visualizing effects in such settings is the heatmap, which plots estimates of the raw methylation effects (as measured by the assay) across subjects using a heat gradient. We implement this in the methyheat function:
methyheat(methyvim_cancer_ate, smooth.heat = TRUE, left.label = "none")
Remark: Figure 4 displays the results of invoking methyheat in this manner produces a plot of the top sites (25, by default) based on the raw p-value, using the raw methylation measures in the plot. This uses the exceptional superheat R package17, to which we can easily pass additional parameters. In particular, we hide the CpG site labels that would appear by default on the left of the heatmap (by setting left.label = "none") to emphasize that this is only an example and not a scientific discovery.
Here we introduce the R package methyvim, an implementation of a general algorithm for differential methylation analysis that allows for recent advances in causal inference and machine learning to be leveraged in computational biology settings. The estimation procedure produces straightforward statistical inference and takes great care to ensure computationally efficiency of the technique for obtaining targeted estimates of nonparametric variable importance measures. A detailed account of the statistical procedure, including an overview of targeted minimum loss-based estimation, is made available in Supplementary File 1. The software package includes techniques for pre-screening a set of CpG sites, controlling for the False Discovery Rate as if all sites were tested, and for visualzing the results of the analyses in a variety of ways. The anatomy of the software package is dissected and the design described in detail. The methyvim R package is available via the Bioconductor project.
methyvim is avaiable on Bioconductor (stable release): https://bioconductor.org/packages/methyvim
Latest source code (development version): https://github.com/nhejazi/methyvim
Archived source code as at time of publication: https://dx.doi.org/10.5281/zenodo.140129818
Documentation (development version): https://code.nimahejazi.org/methyvim
Software license: The MIT License, copyright Nima S. Hejazi
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)