Keywords
Cardiovascular disease, cancer, heart, cardioprotection, cardiotoxicity, prevention, biomarkers
Cardiovascular disease, cancer, heart, cardioprotection, cardiotoxicity, prevention, biomarkers
Recent advances in cancer prevention and management have led to an exponential increase of cancer survivors worldwide1. Regrettably, cardiovascular disease (CVD) has risen in the aftermath as one of the most devastating consequences of cancer therapies2,3, being most prevalent in adult survivors of breast cancer and hematological malignancies1,4,5.
In this work, we define cancer therapeutics-induced cardiotoxicity (CTIC) as the direct or indirect cardiovascular injury or injurious effect caused by cancer therapies, such as mediastinal radiotherapy6 and/or some chemotherapeutic agents7. These incipient toxic changes (e.g. cardiomyocyte apoptosis, cardiac ion-channel alteration, endothelial damage, etc.) can further develop into complex cardiovascular conditions, such as heart failure (HF), valvular heart disease, coronary artery disease (CAD), pericardial disease, systemic and pulmonary hypertension, arrhythmias, and thromboembolic disease, among others8,9. Concomitant pre-existent cardiovascular risk factors have been shown to foment this pathogenesis10.
Doxorubicin (and other agents in the anthracycline family) is the archetype chemotherapeutic leading to CTIC, historically called anthracycline-induced cardiotoxicity or anthracycline-induced cardiomyopathy (AIC)11. The hallmark of this condition is a HF syndrome arising from dilated cardiomyopathy (DCM)11; supraventricular and ventricular arrhythmias have also been described during anthracycline administration but seldom require intervention12. Its prevalence has not been thoroughly studied owing to lack of a uniform definition, inconsistent diagnostic criteria, and underreporting; in modern times, it is thought to affect 17–23% of survivors of pediatric hematological malignancies13–15 and accounts for 2.6% of all patients with non-ischemic cardiomyopathy undergoing cardiac transplantation16.
In addition to anthracyclines, an increasing number of chemotherapeutic agents have been labeled as “cardiotoxic”, with particular mechanisms of action that lead to distinctive cardiovascular effects, and in turn various degrees of frequency and severity (see Table 1 for a list of the most important cardiotoxic chemotherapeutic agents currently available in the US)7,8,17. Because historical cardiotoxicity was mediated by non-specific agents such as anthracycline and alkylating agents, it was believed that the novel “targeted therapeutics” (e.g. monoclonal antibodies, tyrosine kinase inhibitors, etc.) would provide fewer off-target adverse effects. However, an increasingly systematic evaluation and reporting of cardiovascular safety, along with a concomitant explosion of basic18, translational19, and clinical research in the area of CTIC20, have progressively revealed that a large number of these targeted agents are mechanistically determined to cause cardiotoxicity21. Based on the weight of the evidence, the US Food and Drug Administration has recently issued several cardiovascular box warnings for some of these agents, such as myocardial toxicity for anthracyclines, cardiomyopathy for ERBB2 inhibitors, QT prolongation and sudden cardiac death for certain tyrosine kinase inhibitors, and immune-mediated adverse reactions (i.e. myocarditis) for CTLA-4 inhibitors, among others (see Table 1)17.
Text in bold represents US Food and Drug Administration box warnings. 5-FU, 5-fluorouracil; ALK, anaplastic lymphoma kinase; CSF-1R, colony-stimulating factor 1 receptor; ECG, electrocardiogram; EGFR, epidermal growth factor receptor; FKBP, FK506-binding protein; FGFR, fibroblast growth factor receptor; FLT3, FMS-like tyrosine kinase 3; GIST, gastrointestinal stromal tumor; GVHD, graft-versus-host disease; LT3, Lymphotoxin 3; HDAC, histone deacetylase; HGFR, hepatocyte growth factor receptor; HIF-1, hypoxia-inducible factor-1; Ig, immunoglobulin; IGF-1R, insulin-like growth factor 1-receptor; IL, interleukin; LAK, lymphokine-activated killer; mTOR, mammalian target of rapamycin; NK, natural killer; PD-1, programmed death 1; PDGFR, platelet-derived growth factor receptor; PD-L1, programmed death ligand 1; PNET, primitive neuroectodermal tumor; SCD, sudden cardiac death; TdP, Torsades de Pointes; TIL, tumor-infiltrating lymphocyte; VEGF; vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
The significant delay between exposure to mediastinal radiotherapy and manifestation of heart disease, reporting bias, and the frequent concomitant use of cardiotoxic chemotherapy precludes an accurate determination of the incidence of radiation-induced cardiotoxicity8. Having said that, it is believed that cancer survivors who have undergone chest radiotherapy have a 23% increase in absolute risk of cardiovascular morbidity and mortality after 20 years22. When considering the risk of radiotherapy-induced cardiomyopathy, for example, Hodgkin lymphoma survivors who received mediastinal radiotherapy have a fivefold increase after 30 years23, whereas the greatest risk for breast cancer survivors belongs to those who received left-sided chest radiation and concomitant anthracycline chemotherapy24. This laterality risk factor is likely related to the higher incidence of severe CAD in the mid and distal left anterior descending and distal diagonal arteries that is also present in this population, which could contribute to left ventricular (LV) dysfunction25.
Myocardial injury induced by radiotherapy has the hallmark of increased interstitial myocardial fibrosis6, which in turn leads to diastolic LV dysfunction26 and subtle contractile impairment27. These pathological changes may also account for the higher incidence of conduction abnormalities, cardiovascular autonomic dysfunction, impaired exercise performance, and overall mortality28. Additionally, cardiac radiation is associated with complex stenotic and regurgitant valvular lesions29, pericardial disease6, and carotid artery disease30, among other conditions.
Patterned after an established classification of disease progression31, we have divided CTIC into four distinct stages, i.e. A, B, C, and D (see Figure 1). Stage A CTIC refers to cancer patients with cardiovascular health. Stage B CTIC designates cancer patients with high risk of developing CTIC. Risk factors for CTIC can be broadly divided into those pertaining to the patient and those pertaining to the cancer therapies implemented (see Table 2)5,30,32,33. Stage C CTIC denotes “incipient” cardiotoxicity; this is the early stages of the cardiotoxic process before it becomes clinically apparent. This stage is characterized by the appearance of abnormal biomarkers that precede the clearly defined diseased entities (e.g. QTc prolongation precedes Torsade de Pointes and sudden cardiac death, and sarcomeric protein or natriuretic peptide serum elevations precede LV dysfunction and overt heart failure, etc.). Finally, stage D CTIC refers to established cardiotoxicity, which is manifested by cardiovascular syndromes in early or late stages, that requires standard diagnostic modalities and medical and surgical therapies derived from expert consensus guidelines8,31,34–36.
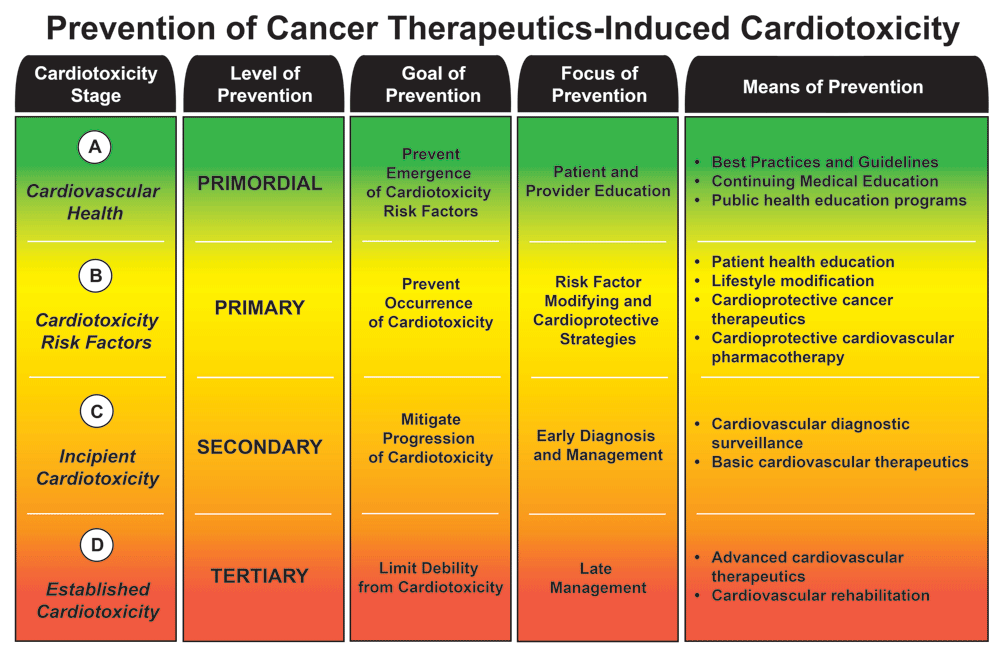
Prevention of cancer therapeutics-induced cardiotoxicity.
CAD, coronary artery disease; CVD, cardiovascular disease; HF, heart failure; LVEF, left ventricular ejection fraction; RT, radiotherapy; SCD, sudden cardiac death; US FDA, United States Food and Drug Administration.
Preventive strategies for CTIC can also be divided into four standard levels, i.e. primordial, primary, secondary, and tertiary, which correspond with the stages of CTIC; each level of prevention has a particular goal, focus, and means (see Figure 1).
Primordial prevention is principally focused on the education of both patients and providers and on the implementation of general best practices to impede the emergence and development of risk factors for CTIC. This is being accomplished by the explosion of expert consensus guidelines in the last decade (see “expert consensus guidelines” below) as well as a growing presence of cardio-oncology programs in major oncology and cardiology scientific meetings. Moreover, there has been an increasing number of continuing medical education materials and public health education programs in this topic, all serving to raise awareness and educate on the cardiovascular effects of cancer therapies. Furthermore, the International Cardio-Oncology Society and the Canadian Cardiac Oncology Network have recently partnered in the writing of a cardio-oncology multidisciplinary training proposal to formally educate physicians in this developing field37.
Primary prevention has the goal of impeding the emergence of CTIC. The diagnosis and control of modifiable risk factors (see Table 2) and the promotion of cardiovascular health in the cancer population are of utmost importance. In addition, the administration of cardioprotective therapies to selected patients with unavoidable moderate and high risk of CTIC is a means of primary prevention (see “cardioprotectants” below).
Secondary prevention is enforced once cardiac toxicity is incipient; early diagnosis and surveillance (see “blood biomarkers and diagnostic modalities” below), implementation of cardioprotective strategies, and administration of cardioprotective and basic therapies have the overarching goal to mitigate the progression of cardiotoxicity, restore cardiovascular health, and prevent complications. As in most health conditions, earlier diagnosis and treatment of CTIC seem to translate into improved outcomes38. Inspired by the American Society of Clinical Oncology (ASCO) clinical practice guideline on the prevention and monitoring of cardiac dysfunction in survivors of adult cancers5, as well as by other recent expert consensus guidelines that include recommendations on the prevention of CTIC8,32,39,40, we have constructed a table summarizing the general evidence-based recommendations for the prevention of cardiotoxicity before, during, and after cancer therapies (see Table 3).
DM, diabetes mellitus; HL, hyperlipidemia; HTN, hypertension.
Lastly, once CTIC has progressed sufficiently to be manifest in cardiovascular syndromes (e.g. HF, arrhythmias, acute coronary syndromes, etc.), tertiary prevention aims to limit further progression and disability, and promote rehabilitation, by both basic and advanced cardiovascular therapeutics. The evaluation and management of these defined CTIC syndromes are similar to those encountered in non-cancer patients. There are several clinical practice guidelines for the evaluation and management of these conditions in the literature31,35,36,41,42, and some specifically address the cancer population8,9; these tertiary prevention strategies will not be further detailed in this work.
As mentioned above, the prevention of cardiotoxicity induced by cancer therapies has increasingly been the focus of several clinical cardiovascular and oncological societies, demonstrating the increasing relevance that this field has taken in the latest decade. In 2012, the European Society for Medical Oncology published a basic set of clinical practice guidelines for the prevention, monitoring, and management of CTIC40. The American Society of Echocardiography and the European Society of Cardiovascular Imaging joined forces to create expert consensus guidelines for the multimodality imaging evaluation of cardiovascular complications of radiotherapy in adult patients in 201330 as well as evaluation during and after cancer therapies in 201443. These efforts aim to standardize the indications, acquisition protocols, definitions, limitations, and vendor variability for the different cardiac imaging modalities usually employed in the diagnosis and surveillance of CTIC. In 2016, the American Heart Association (AHA) released a comprehensive scientific statement describing the mechanism, magnitude, onset, and likelihood of direct myocardial toxicity of several anti-cancer medications, among other clinically approved drugs, “to assist healthcare providers in improving the quality of care for these patients”7. In the same year, the Canadian Cardiovascular Society published a set of best practice guidelines for the management of cancer patients, focusing on the identification of the high-risk population and the detection and prevention of cardiotoxicity39. This was followed by a position paper from the European Society of Cardiology summarizing the available evidence on the pathophysiology, prevention, diagnosis, therapeutic management, and long-term surveillance of the most common forms of cardiotoxicities induced by cancer therapies8. Most recently, as mentioned above, the ASCO published a clinical practice guideline outlining general recommendations for the prevention of cardiac dysfunction in survivors of adult cancers5. It was developed by an expert multidisciplinary physician panel using a systematic review (1996–2016) of 104 articles (meta-analyses, randomized clinical trials, and observational trials) and their clinical experience. Finally, the AHA has just published a scientific statement specifically and comprehensively dealing with the prevention of CVD in breast cancer patients, including that caused by cancer therapies32.
The development and investigation of cardioprotective agents has been exponentially increasing since the early days of anthracycline cardiotoxicity. To date, only one cardioprotectant is approved for clinical use, i.e. dexrazoxane; many others have been tested in the clinical setting, and an even larger number are on preclinical stages of investigation (see Table 4 for a succinct list of cardioprotective agents for CTIC that have been shown to be useful at different stages of research). The vast majority of cardioprotectants have been tested in the setting of anthracycline administration, either alone or in combination with other chemotherapeutic agents; a small number has been tested in trastuzumab-only administration.
ACEI, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; MRA, mineralocorticoid receptor antagonist; NSAID, non-steroidal anti-inflammatory drug; PC-SOD, lecithinized human recombinant super oxide dismutase.
| Cardioprotectants in cancer therapeutics-induced cardiotoxicity | |||
|---|---|---|---|
| Clinical | Antidotes | Dexrazoxane | Lipshultz et al.49 |
| N-acetylcysteine | Myers et al.54 | ||
| Beta-blockers | Carvedilol | Avila et al.55 | |
| Nebivolol | Kaya et al.56 | ||
| Bisoprolol | Pituskin et al.57 | ||
| Metoprolol | Georgakopoulos et al.58 | ||
| ACEIs | Enalapril | Cardinale et al.59 | |
| Ramipril | Jensen et al.60 | ||
| Perindopril | Pituskin et al.57 | ||
| ARBs | Valsartan | Nakamae et al.61 | |
| Candesartan | Gulati et al.62 | ||
| MRAs | Spironolactone | Akpek et al.63 | |
| Statins | Atorvastatin | Acar et al.64 | |
| Natural supplements | Melatonin | Lissoni et al.65 | |
| Ubiquinone | Iarussi et al.66 | ||
| Vitamins C and E | Wagdi et al.67 | ||
| Levocarnitine | Waldner et al.68 | ||
| Preclinical | ACEIs | Temocapril | Tokudome et al.69 |
| Delapril | Maeda et al.70 | ||
| Zofenopril | Sacco et al.71 | ||
| ARBs | Losartan | Matouk et al.72 | |
| Statins | Fluvastatin | Riad et al.73 | |
| Biguanides | Metformin | Kobashigawa et al.74 | |
| Prostacyclins | Iloprost | Neilan et al.75 | |
| NSAIDs | Meloxicam | Hassan et al.76 | |
| Vasodilators | Diazoxide | Hole et al.77 | |
| Molsidomine | Disli et al.78 | ||
| Nicorandil | Ahmed et al.79 | ||
| Iron salts | Ferric carboxymaltose | Toblli et al.80 | |
| Neuropeptides | Ghrelin | Wang et al.81 | |
| Natural antioxidants | Dihydromyricetin | Zhu et al.82 | |
| Hydroxytyrosol | Granados-Principal et al.83 | ||
| Sesame oil | Saleem et al.84 | ||
| Sesamin | Su et al.85 | ||
| Salidroside | Wang et al.86 | ||
| Glutathione | Mohamed et al.87 | ||
| Quercetin | Matouk et al.72 | ||
| Isorhamnetin | Sun et al.88 | ||
| Cannabidiol | Fouad et al.89 | ||
| Resveratrol | Dolinsky et al.90 | ||
| indole-3-carbinol | Hajra et al.91 | ||
| α-Linolenic acid | Yu et al.92 | ||
| Synthetic antioxidants | Didox | Al-Abd et al.93 | |
| Other | Mdivi-1 | Gharanei et al.94 | |
In the US, dexrazoxane is the only approved cardioprotective agent consistently shown to reduce the incidence or severity of AIC44. It is recommended to be given intravenously, in a 10:1 ratio of dexrazoxane:doxorubicin (e.g. dexrazoxane 500 mg/m2:doxorubicin 50 mg/m2) in the context of normal renal function; cardiac monitoring should be continued during dexrazoxane therapy17. Its use has been associated with statistically significant risk reductions for most doxorubicin-related cardiotoxic outcomes (other than survival)45, without compromising its therapeutic efficacy, in both pediatric and adult populations46–49. Although currently dexrazoxane use is strictly restricted to women with metastatic breast cancer who have received a cumulative doxorubicin dose of 300 mg/m2 and need continued treatment to maintain tumor control44,50, its use in the treatment of other malignancies has been endorsed by expert guidelines51. Having said that, dexrazoxane is not currently recommended for routine use with the initiation of doxorubicin therapy for either primary or metastatic disease51–53. It needs to be noted that dexrazoxane was associated with a potential increased risk of acute myeloid leukemia, myelodysplastic syndrome, and second malignant neoplasms in a pediatric population with Hodgkin lymphoma in a single study a decade ago95. Many later studies have not been able to reproduce these initial results45,96–98. Furthermore, a recent large clinical trial in a pediatric population corroborated these latter findings, suggesting that dexrazoxane was indeed cardioprotective, did not interfere with antitumor efficacy, did not result in an increased occurrence of toxicities, and had no association with a significant rise in second malignancies99.
Given their consistent benefit in other cardiovascular conditions (e.g. HF and CAD), beta-blockers, angiotensin converting-enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), mineralocorticoid receptor antagonists (MRAs), and HMG-CoA reductase inhibitors (statins) have been extensively studied in the clinical setting, in the context of both anthracycline and trastuzumab therapy, for the prevention of LV dysfunction.
Beta-blocker agents with antioxidant properties such as carvedilol100–102 and nebivolol56 have shown the most promising results in early small clinical trials investigating their cardioprotective effects. Regrettably, in the so far largest clinical trial of beta-blockers for the prevention of cardiotoxicity under contemporary anthracycline dosage, carvedilol monotherapy had no impact on the incidence of early onset of LV ejection fraction (LVEF) reduction when compared to placebo in a breast cancer population55. Similarly, ACEI monotherapy with enalapril59 and ramipril60 has also been shown to be beneficial in early small clinical trials; however, the administration of enalapril monotherapy either before chemotherapy or during or after chemotherapy in selected patients with elevated serum troponin levels failed to have a significant impact on outcomes in the most recent multicenter clinical trial103. As for ARBs, valsartan was shown to be beneficial in small clinical trials over a decade ago61; however, the use of candesartan as a cardioprotectant has recently provided conflicting results in well-conducted randomized placebo-controlled clinical trials104,62. The cardioprotective effects of spironolactone monotherapy have also been promising in early small clinical settings63, but data from larger randomized clinical trials are still lacking.
Several clinical trials have investigated the cardioprotective effects of combined neurohormonal inhibition, i.e. beta-blockers plus ACEIs/ARBs, as is recommended in the general population with HF34. Over a decade ago, early initiation of combined beta-blockers and ACEIs was shown to provide benefit in a small population of established AIC, albeit the effect was thought to be mediated mainly by beta-blockers105. Since then, the role of combined neurohormonal inhibition in cardioprotection has been repeatedly evaluated up to this day in the settings of anthracycline, trastuzumab, or sequential chemotherapy. In the only positive trial to date, the combination of enalapril and carvedilol was shown to prevent deterioration of LV function in adult patients with hematological malignancies undergoing anthracycline therapy106. However, there are significant concerns regarding this trial, including lack of blinding and differing results based on the methods used to quantify LVEF, making it difficult to conclusively interpret107. In other clinical settings, metoprolol has been tested in combination with enalapril58 and with candesartan62, with disappointing results. Similarly, the combination of bisoprolol and perindopril failed to prevent trastuzumab-induced LV remodeling in a modern cohort of ERBB-positive breast cancer patients57. Finally, in the as-yet-unpublished work by Guglin et al. presented at the 2018 American College of Cardiology annual meeting, both lisinopril and carvedilol failed to prevent cardiotoxicity in breast cancer patients treated with trastuzumab monotherapy, whereas both drugs prevented cardiotoxicity in patients who received both anthracycline and trastuzumab sequential therapy108.
The cardioprotective role of statins has also been evaluated in small retrospective and prospective analyses, both with non-specific statins109,110 and atorvastatin monotherapy64, and was found to be beneficial. These findings are very promising but are yet to be corroborated in larger randomized placebo-controlled trials (simvastatin NCT02096588; atorvastatin NCT02674204).
Clinical cardioprotective data involving natural supplements are scarce but growing. Ubiquinone (coenzyme Q10) administration in children receiving anthracyclines was associated with a lesser degree of LV dysfunction and remodeling66. N-acetylcysteine, administered either alone or with vitamins E and C, averted LV dysfunction from developing in patients receiving high-dose doxorubicin and/or radiotherapy, respectively67,54. Melatonin65 and levocarnitine68 have also been tested in the clinical setting with positive results. Larger randomized placebo-controlled trials are lacking as to draw firm conclusions relevant to the clinical practice.
Many other agents have been shown to ameliorate anthracycline cardiotoxicity in small animal models of CTIC. Clinically available agents such as losartan72, fluvastatin73, metformin74, iloprost75, and meloxicam76 as well as other clinically unavailable ACEIs69–71 have been shown to have cardioprotective results in vivo. Vasodilators77–79, neuropeptides81, and iron salts80 have also been found to be useful. Finally, given that the pathogenesis of anthracyclines is in part related to increased oxidative stress100, several natural antioxidants (e.g. sesamin85 and sesame oil84 and hydroxytyrosol83, among others82,86–92) have been tested and shown various degrees of cardioprotective effects. Didox, a synthetic antioxidant, was also shown to significantly potentiate the cytotoxicity of doxorubicin in liver cancer cells while at the same time protecting the murine model from cardiotoxicity93. Mdivi-1, a mitochondrial division/mitophagy inhibitor, was also shown to lessen AIC94.
Within a family of cardiotoxic agents, there are variations in terms of cardiac safety. For example, the use of pegylated liposomal doxorubicin has been associated with a lower incidence of CTIC and HF111,112. Similarly, epirubicin or mitoxantrone are also believed to cause less cardiotoxicity compared with doxorubicin113. When considering the large family of multitargeted tyrosine kinase inhibitors, vandetanib, nilotinib, and ponatinib seem to possess the highest cardiotoxicity risk17. The role of exercise therapy in the prevention of CTIC remains controversial because of conflicting results114,115.
In summary, with the exception of dexrazoxane, no conclusive recommendations can be made on the clinical use of cardioprotectants for either stage B or stage C CTIC5.
Blood biomarkers, in particular myocardial natriuretic peptides (i.e. NTproBNP and BNP) and sarcomeric proteins (i.e. troponin I and T), have been an integral part of the diagnostic and prognostic armamentarium in common cardiovascular conditions, such as HF and CAD. As it would seem natural, they have been progressively adopted in clinical practice to assist in the diagnosis or surveillance of patients with incipient and established CTIC, in particular LV dysfunction and HF (see Table 5 for a list of various clinical and preclinical biomarkers shown to predict CTIC)5.
ANP, atrial natriuretic peptide; BNP, B-type natriuretic peptide; cMLC1, cardiac myosin light chain-1; cTnAAbs, cardiac troponin specific autoantibodies; cTnI, cardiac troponin I; cTnT, cardiac troponin T; GWAS, genome-wide association study; hs-CRP, high-sensitive C-reactive protein; hs-TnI, high-sensitive troponin I; GDF15, growth differentiation factor-15; GPBB, glycogen phosphorylase BB; IMA, ischemia modified albumin; MPO, myeloperoxidase; NTproBNP, amino-terminal pro B-type natriuretic peptide; PlGF, placental-derived growth factor; ROS, reactive oxygen species.
| Blood biomarkers in cancer therapeutics-induced cardiotoxicity | |||
|---|---|---|---|
| Clinical | Myocardial natriuretic peptides | NTproBNP | De Iuliis et al.119 |
| BNP | Lenihan et al.37 | ||
| ANP | Nousiainen et al.120 | ||
| Myocardial sarcomere proteins | cTnI | Cardinale et al.117 | |
| cTnT | Kilickap et al.118 | ||
| hs-cTnI | Sawaya et al.121 | ||
| hs-cTnT | Katsurada et al.122 | ||
| us-cTnI | Ky et al.123 | ||
| Other biomarkers | cTnAAbs | Ylänen et al.124 | |
| Hb | Garrone et al.125 | ||
| hsCRP | Onitilo et al.126 | ||
| MPO | Ky et al.123 | ||
| PIGF | Putt et al.127 | ||
| GDF15 | Arslan et al.128 | ||
| Arginine-NO metabolites | Finkelman et al.129 | ||
| GPBB | Horacek et al.130 | ||
| ROS | Mercuro et al.131 | ||
| IMA | Ma et al.132 | ||
| Single nucleotide polymorphims (GWAS) | rs2229774 | Aminkeng et al.133 | |
| rs1786814 | Wang et al.134 | ||
| rs28714259 | Schneider et al.135 | ||
| Preclinical | DNA | Doxorubcin DNA adducts | Hahm et al.136 |
| Spp1, Fhl1, Timp1, Ccl7 and Reg3b | Mori et al.137 | ||
| MicroRNA | miR-34a | Desai et al.138 | |
| miR-34c | Vacchi-Suzzi et al.139 | ||
| miR-146a | Horie et al.140 | ||
| Proteins | S100A1 | Eryilmaz et al.141 | |
| cMLC1 | ElZarrad et al.142 | ||
| Cathepsin B | Bao et al.143 | ||
| Proteomics pattern diagnostics | Petricoin et al.144 | ||
| Metabolomics pattern diagnostics | Li et al.145 | ||
| Transcriptome profiling | Todorova et al.146 | ||
Troponin I59,116,117 and troponin T118 have been shown to be clinically useful in several clinical trials of cardiotoxicity prediction. Modern, more-sensitive assays of troponin I and T (high-sensitivity and ultra-sensitivity) have also been shown to be clinically predictive of CTIC121–123. Early studies have suggested that troponin I elevation predicted severity of CTIC116,117, and refractoriness to HF therapy in the case of trastuzumab-induced cardiomyopathy117, but response to enalapril monotherapy in the case of AIC59. However, in a recent large multicenter randomized clinical trial, these findings could not be corroborated103. Interestingly, the presence of troponin-specific autoantibodies also predicted cardiac dysfunction by cardiac magnetic resonance (CMR) imaging in the absence of elevated traditional troponin levels124. Myocardial natriuretic peptides, such as NTproBNP119, BNP147, and ANP120, have also been shown to be clinically useful predictors of CTIC, albeit to a lesser extent.
Although the use of these blood biomarkers is currently recommended in the evaluation and surveillance of patients with CTIC5,8, their helpfulness remains disputed owing to inconsistent results in terms of sensitivity, accuracy, and reliability148. Hence, various other alternative blood biomarkers have been studied in recent years, either alone or in combination, and shown also to be clinically predictive of CTIC, e.g. hsCRP126, MPO123, and arginine-NO metabolites (arginine, citrulline, ornithine, asymmetric dimethylarginine, symmetric dimethylarginine, and N-monomethylarginine)129, among others125,127,128,130–132. Likewise, many other predictive biomarker strategies are currently being developed in the preclinical arena. Proteomics144 and metabolomics145 pattern diagnostics, as well as transcriptome profiling146, have been shown to be useful in animal models of AIC as well as the detection of doxorubicin DNA adducts (HM-dUMP, 8-OH-dGMP, HM-dCMP, and Me-dCMP)136 and other particular genes that are overexpressed during incipient cardiotoxicity137. Cellular proteins such as S100A1141, cMLC1142, and cathepsin B143 have also been shown to have predictive value. Some microRNAs (e.g. miR-34a138, miR-34c139, and miR-146a140) have been shown to be useful in predicting CTIC in small animal models; however, a recent clinical trial involving miR-208a measurement in breast cancer patients failed to have a predictive impact149.
Finally, research efforts to identify the genetic susceptibility of AIC have been increasing in the last decade, with the purpose of risk stratifying patients before they receive anthracycline chemotherapy. To date, three main single-nucleotide polymorphisms (SNPs: rs28714259135, rs1786814134, and rs2229774133) have been identified as being strongly associated with AIC by means of genome-wide association studies (GWAS) from pediatric and adult case-controlled clinical trial populations.
Non-blood diagnostic modalities are also an integral part of the evaluation of CVDs. For the purpose of early diagnosis and surveillance of CTIC, several imaging modalities have been studied since the late 1970s and shown to be of value (see Table 6). Historically, electrocardiography150 was used to diagnose arrhythmias during anthracycline infusion, and radionuclide cineangiography (MUGA)151,152 was the first technique used to detect falls in LV systolic function in patients receiving anthracyclines153. Although MUGA is still considered widely available and highly reproducible, it carries the main disadvantage of submitting cancer patients to small, but potentially significant, radiation exposure (5–10 mSv)30,43. Additionally, 2D-echocardiogram154 and stress 2D-echocardiogram155 have been shown to be beneficial in the serial evaluation of cancer patients undergoing cardiotoxic chemotherapies. Newer echocardiographic modalities, such as 3D-echocardiography156 and LV global longitudinal strain (LVGLS) measurement by speckle-tracking echocardiography (STE)157, have demonstrated superiority over 2D-echocardiography in terms of reproducibility and predictability, respectively. CMR is currently considered the gold standard modality in the assessment of LV and right ventricular volumes and function158. Secondary modalities such as CMR strain imaging159, T1 mapping160, and extracellular volume fraction (ECV)161 have also been clinically studied in recent years and found to be of great value in the assessment of subclinical cardiotoxicity. Among various non-imaging techniques, cardiopulmonary exercise testing was shown to detect abnormalities in peak oxygen consumption in cancer patients with apparently normal LV function162, suggesting subclinical impairments of contractile reserve and chronotropic incompetence28. Finally, many other imaging modalities are currently being studied in the preclinical arena to help detect incipient cardiotoxicity with high specificity and sensitivity. For example, 18F-labeled tetrapeptide caspase positron emission tomography (PET) is able to specifically diagnose doxorubicin-induced myocardial apoptosis in a murine model by detection of overexpressed myocardial caspase 3 resulting from anthracycline chemotherapy163.
2D, two-dimensional; 3D, three-dimensional; 99m Tc, technetium-99; CMR, cardiac magnetic resonance; CPET, cardiopulmonary exercise testing; ECG, electrocardiogram; ECV, extracellular volume fraction; LVEF, left ventricular ejection fraction; LVGLS, left ventricular global longitudinal strain; MUGA, multigated acquisition; PET, positron emission tomography; RBC, red blood cells.
| Diagnostic modalities in cancer therapeutics-induced cardiotoxicity | ||
|---|---|---|
| Established clinical | ECG | Steinberg et al.150 |
| MUGA (99m Tc-labeled RBC) | Schwartz et al.151 | |
| Stress MUGA | McKillop et al.152 | |
| 2D-echocardiography | Thavendiranathan et al.154 | |
| Stress 2D-echocardiography | Khouri et al.155 | |
| CPET | Jones et al.162 | |
| Novel clinical | 3D-echocardiography | Walker et al.156 |
| Speckle-tracking echocardiography (LVGLS) | Negishi et al.157 | |
| CMR | Armstrong et al.158 | |
| CMR strain imaging | Drafts et al.159 | |
| CMR T1 mapping | Lightfoot et al.160 | |
| CMR ECV | Jordan et al.161 | |
| Preclinical | PET (18F-labeled tetrapeptidic caspase) | Su et al.163 |
According to current guidelines, echocardiography (ideally 3D-echocardiography) is the method of choice for the evaluation of patients before, during, and after cancer therapies43. CMR and MUGA scan (in that order) should be utilized as alternative modalities whenever the echocardiographic image quality is deficient5. When available, measurement of LVGLS by STE is also recommended as a complementary modality5. CMR should also be considered for the evaluation of chronic “constrictive” pericarditis, when the diagnosis remains uncertain after a careful echocardiographic evaluation43.
To date, there is little evidence to guide the indication, timing, and frequency of use of imaging modalities in patients undergoing cancer therapies. The ASCO expert consensus recommends an echocardiographic evaluation prior to the initiation of potentially cardiotoxic cancer therapies5. Routine imaging surveillance in asymptomatic patients should be offered to patients based on the healthcare provider’s perceived risk of CTIC, and the frequency of it needs to be individualized based on clinical judgment and patient circumstances5. Subsequent to cardiotoxic cancer therapies, it is recommended that high-risk patients undergo a follow up LVEF evaluation between 6 and 12 months after completion of therapy5.
In this work, we have attempted to comprehensively and concisely survey the most relevant available literature pertaining to cardioprotection during cancer therapy. We have briefly summarized the pathophysiology of CTIC, describing the mechanisms of cardiotoxicity of various agents, and risk factors that promote this phenomenon. For didactic purposes, we have classified CTIC into four progressive stages, in which four levels of prevention are applied, each having a specific goal, focus, and means of prevention. We have subsequently reviewed the available data on cardioprotective agents, blood biomarkers, and imaging diagnostic modalities, which are the core of primary and secondary prevention strategies. Finally, we have provided general evidence-based preventive recommendations for CTIC following the most current expert consensus guidelines. The promotion of the cardiovascular health of cancer patients and cancer survivors is paramount, requiring the diligent and knowledgeable effort of a multidisciplinary team of healthcare providers; as in all medical disorders, prevention is better than cure.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)