Keywords
Tar, Sand, Microbiota
Tar, Sand, Microbiota
‘Tar sands’ is a term used to describe a combination of bituminous sand, sandstone, oil-impregnated rock, oil sand and rock asphalt1. The tar sands is a rapidly developing source of unconventional petroleum. The bitumen, an oil-rich residue, can be extracted from the tar sands and refined into crude oil. In the United States, occurrences of tar sands have been reported in 22 states; however, one of the largest deposit is located in southwestern Utah, with estimated recoverable oil ranging from 12 to 20 barrels2.
It has been suggested that tar sands were created by the microbial degradation of immobile subsurface oil over several million years3. Little is known about the origin of the microorganisms in the tar sands of Utah, although the indigenous microbial communities exist in extreme conditions in the bituminous tar sands and are limited by harsh conditions such as low moisture and oxygen, recalcitrant hydrocarbons and a high concentrations of toxic metals4. However, a paucity of data exists on the indigenous microbial communities of the tar sands of Utah.
Traditionally, the diversity of the bacterial communities in the tar sands has been investigated using the isolation and cultivation approaches5. However, only 1.5% of bacteria in the soil can be readily cultured6,7. Most cultural conditions cannot mimic the specific microhabitats that many prokaryotes thrive within. Therefore, new methods for the analysis of whole microbial community structure and metabolic function in the bituminous tar sands have been developed. One of the methods developed to provide a dynamic tool for the assessment of microbial community structure and function is the BIOLOG™ Community Level Physiological Profile (CLPP). This method is based upon the preferential metabolism of 31 different carbon sources on a microtiter plate. These carbon sources include a wide range of chemical classes, such as carbohydrates, esters, polymers, carboxylic acids, alcohols, amides, phosphorylated chemicals, amino acids, aromatic chemicals, and amines8. Each well on the BIOLOG™ Ecoplate contains a unique carbon source, peptone, and a 2,3,5-triphenyltetrazolium chloride (TTC) dye. NADH is produced from the respiration of the specific carbon sources. The final electron acceptor TTC is irreversibly reduced to formazan, a red pigment that can be quantified visually by use of a microplate reader8,9. The intensity of the color change correlates to the amount of metabolism of the carbon source in that well. The net intensity of the color change is calculated by subtracting the absorbance of the non-carbon-source control well. The oxidation of the carbon substrate oxidized by the microbe can be considered to be its metabolic fingerprint. However, Windig10 reported that the BIOLOG™ system excludes strict anaerobes and bacteria that lack certain electron transport enzymes. Yao et al.11 advocated that CLPP is selective and favors microbes that grow quickly or those with a high inoculum density in the initial sample. Hence, culture-independent analysis is the method of choice for the investigation of the bituminous tar sands.
The objective of this study was to identify and characterize the indigenous bacterial communities of the Utah tar sands using next-generation sequencing technology and to assess the functional diversity using the CLPP system. The overall goal of this project is significant because studies have demonstrated that naturally occurring microbes can be harnessed for the degradation of recalcitrant polyaromatic hydrocarbons, heavy metals, and naphthenic acids. The combination of the functional diversity and the characterization of the indigenous microbiota will advance our understanding of the fate of tar-associated potentially toxic compounds by environmental microbiota.
The University of Alberta Geotechnical Centre, Canada, kindly provided the tar sands sample. All samples were shipped on ice to Florida A&M University where they were processed for metagenomics and CLPP analyses. Genomic DNA was extracted from the samples using the PowerSoil DNA Isolation kit (MoBio Laboratories, Inc., Carlsbad, CA, USA). Bar-coding pyrosequencing of the 16S rRNA gene V4 region was performed using the Earth Microbiome Project (EMP) standard protocols (http://www.earthmicrobiome.org/emp-standard-protocols) and sequenced on a Roche 454 FLX instrument (Roche, Indianapolis, IN, USA) with titanium reagents, following manufacturers recommended procedures. Sequences that passed quality controls were uploaded to MOTHUR v.1.33.3, where tags, low-quality sequences and chimeric reads were removed.
CLPP of the tar sand sample was analyzed using BIOLOG™ Ecoplates, as described previously8,12. Triplicate samples of the BIOLOG™ Ecoplate were used for this experiment. A total of three sets of 1 g samples of the tar sand were shaken in 99 ml of distilled sterile water for 20 min at 20°C. The samples were then incubated at 4°C for 30 min and then centrifuged for 10 min at 500g. The supernatant of the tunes were pooled and thoroughly mixed. In total, 150 μl of the supernatant was inoculated into each well of the Biolog EcoPlates. This ensured that each of the 96-well plate contained three replicates of each sample exposed to 31 different carbon sources. Water blanks were included with each plate and the plates were incubated at 25°C without shaking. Substrate utilization was monitored by measuring absorbance at 595 nm using a BIOTEK UQuant Microplate Spectrophotometer, (BIO-TEK Instruments, Inc., USA). The first measurement was made immediately after inoculation and subsequent readings were taken after every 24 h interval for 7 days. The measurements of individual substrates were corrected for background absorbance by subtracting the absorbance of the control (water) samples. If a negative number was obtained, it was manually set to zero. A well was considered positive if the mean OD595nm exceeded that of 0.25.
In this study the 31 carbon sources were organized into groups 1–5 as described by Zak et al.13. Carbon sources originally grouped as miscellaneous by Zak et al.13 or those new to the BIOLOG ™ EcoPlate, were grouped into one of the other five categories - carbohydrates, carboxy acids, amino acids, esters and polymers. Grouping the data into 5 guilds compresses a 31-dimensional data set into 5 dimensions, significantly reducing the complexity of the data and subsequent interpretation14.
The average well color development (AWCD) was used to assess the microbial response for all the carbon sources. The AWCD was determined as follows:
where ODi is optical density value from each well, corrected subtracting the blank well (inoculated with water) values from each plate well8,15. The 96-h data was used for statistical analysis of CLPPs.
The diversity of substrate utilization was calculated using the Shannon’s diversity index (H) and evenness (E)16. The Shannon-Weaver index is a measure of the capacity of the bacterial community to degrade the carbon sources in the well and can be considered as an index of the physiological diversity of the bacterial community in the tar sands. Higher values of H indicate the ability of the microbial community to degrade the substrates with a high efficiency16:
where Pi = (OD reading of well I)/(sum of all wells)], based on the OD in the Ecoplates.
The Shannon evenness (E) is a measure of how dissimilar the abundances of the species in a community are from each other17 and was calculated as:
E – H/lnS
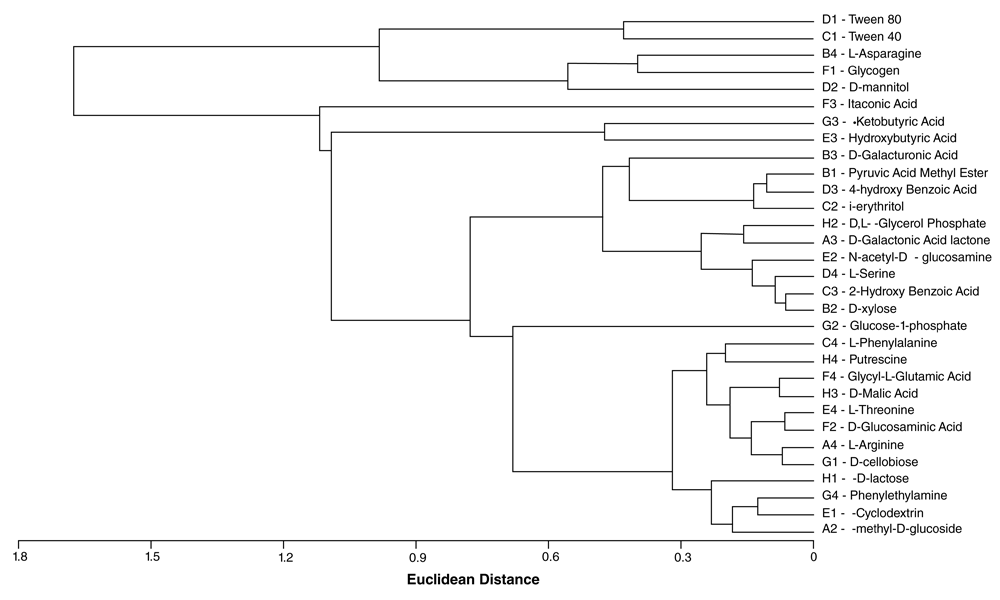
where S (Shannon richness) is the number of substrates used by the microbial communities. Further, the cluster analysis of the substrate utilization pattern was constructed using the nearest neighbor method with Euclidian distance to form linkage dendrogram.
In this study, the compositions of bacterial community of the tar sands was investigated by a pyrosequencing-based analysis of the 16S rRNA gene sequences. The gram-positive Actinobacteria (50%) were dominant in the tar sand followed by Betaproteobacteria (27%), Alphaproteobacteria (7%), Gammaproteobacteria (7%) and Acidobacteria (2%) (Figure 1). Actinobacteria are known for their role in the biodegradation of a variety of different pollutants including petroleum hydrocarbons18,19. The predominant genus identified in these tar sands was Arthrobacter, followed by Dietzia, Janibacter, Nocardioides, Microbacterium, Agrococcus and Salinibacterium (Figure 2). Most of these genera have also been previously shown to degrade aromatic hydrocarbons, indicating that tar sands are a very active repository of hydrocarbonoclastic microorganisms. When the taxonomic affiliation of the obtained metagenomic sequences were investigated, we found that the gram-positive Arthrobacter spp. from the Actinobacteria phyla were predominant, which comprised approximately half of the total microbial community assemblage in this particular Utah tar sand. Arthrobacter species possess a significant hydrocarbonoclastic potential as demonstrated by previous studies and we recommend that Actinobacteria, native to the tar sand habitats, should be targeted for future research on this area.
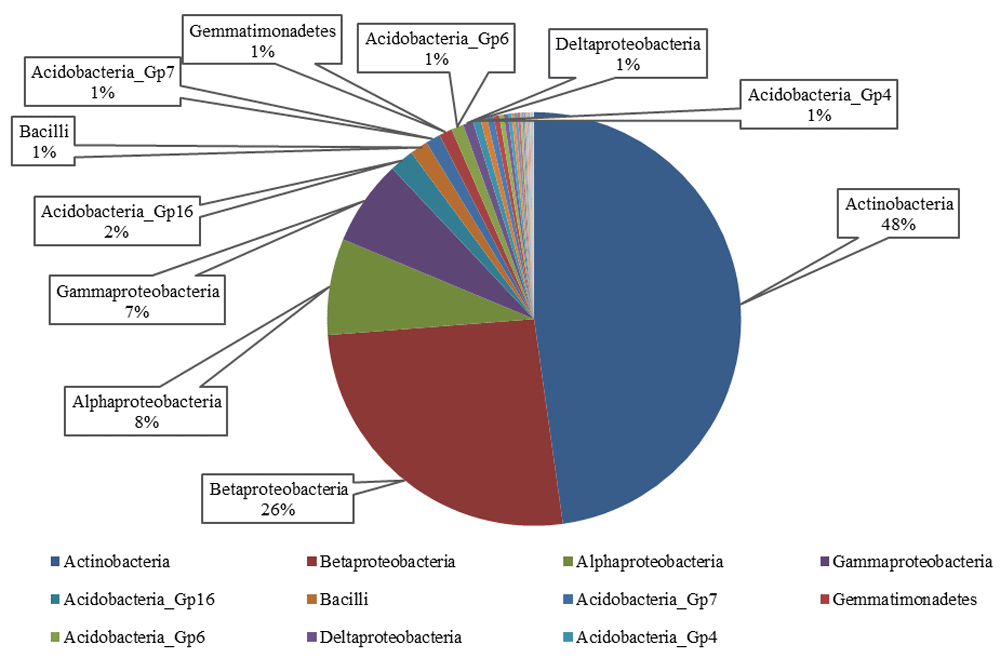
The gram-positive Actinobacteria were dominant in the tar sand followed by Betaproteobacteria, Alphaproteobacteria, Gammaproteobacteria and Acidobacteria.
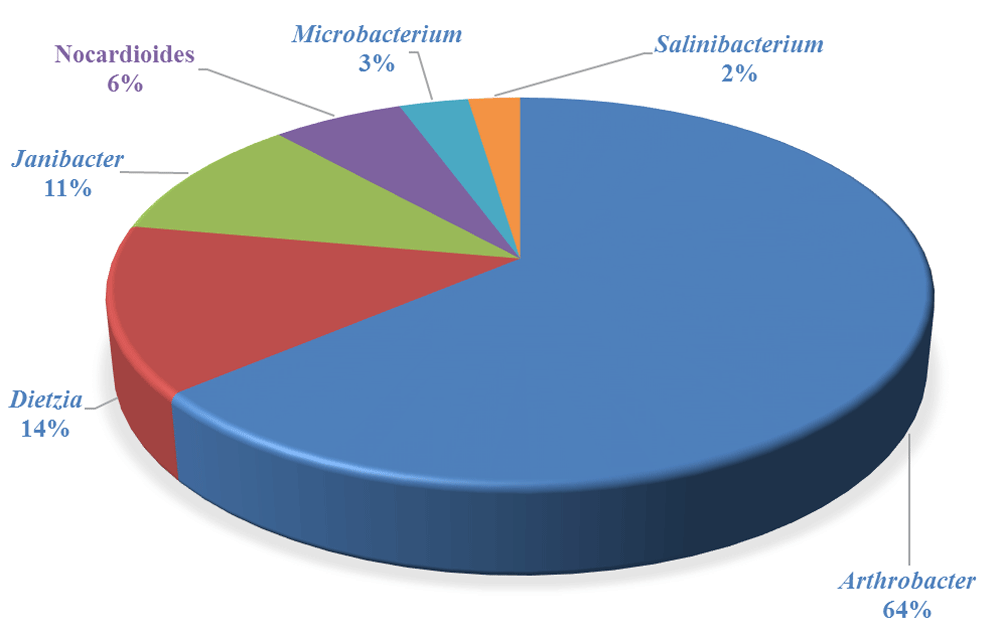
The gram-positive Arthrobacter spp. from the Actinobacteria phyla were dominant in the tar sand microbiota.
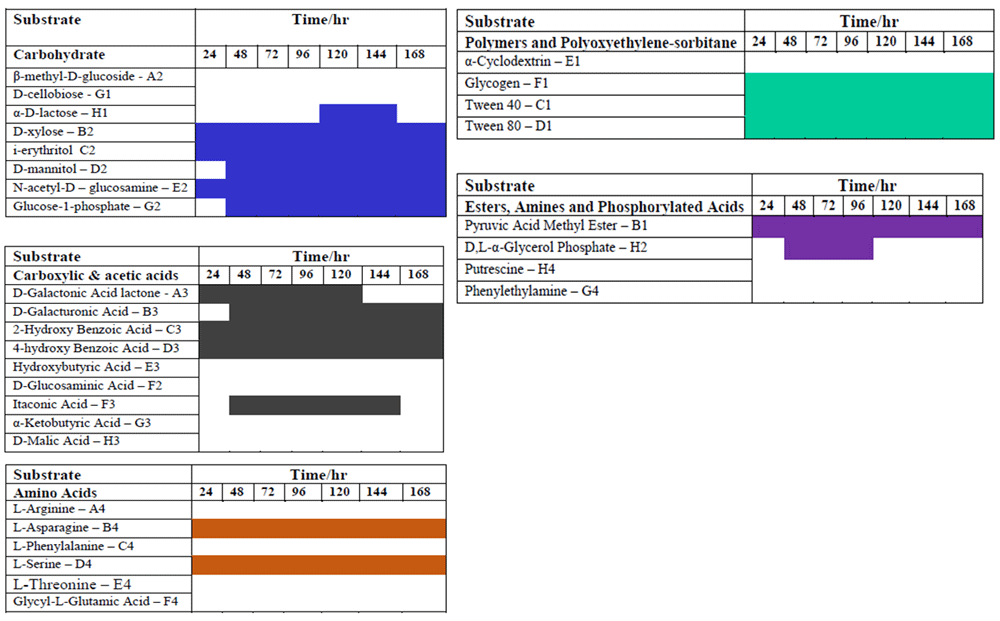
Biolog® Ecoplates were used to evaluate the functional diversity of the microbial community of the tar sands. The AWCD revealed sigmoidal relationships between the OD590 and time for all carbon sources (polymers, carbohydrates, amino acids, esters) except the carboxylic acids (Figure 3). This pattern for the polymers, carbohydrates, amino acids, and esters is fundamentally similar to the bacterial growth curve. Yao et al20 suggested that color development reflected species metabolic activity and the ability of the bacterial community to respond to substrates. The AWCD for all the substrates in the Biolog® Eco-microplates was 0.25. Less than 24h was needed for the first color development the microplate wells containing the polymers whereas the duration of the lag phase of the carboxylic acids was prolonged. The first color development of the carboxy acids (AWCD0.2) was recorded at 48 h (Figure 3). The highest utilization was observed in the microplate wells containing the polymers (AWCD0.78) after 96 hours. The observed differences in the substrate utilization pattern of five major carbon groups (Figure 3) might be due to the presence of different functional groups, such as carbohydrates R-C=O), amino acids (-NH2 and -COOH), carboxylic acid (-COOH), esters, amines and phosphorylated acids (-COOR’, -NH2), and polymers (–(CH2-CH2)–(R)n). The high utilization of the recalcitrant polymers suggest that the polymers could be more easily utilized by the indigenous microbial community in the tar sands. After 96 h, there was a decrease in the rate of AWCD; thus, the analysis of functional diversity of microbial community of tar sands was completed at 96 h. The decreased rate may be due to the change of metabolic activity of active microbial communities utilizing the C-substrates. Cluster analysis of the substrates revealed a systematic grouping of the substrates based on their utilization pattern (Figure 4).
The Shannon-Wiener index is an indication of the spread or distribution of carbon source utilization by the microbial community21. Typical values of the Shannon-Wiener Index usually lies between 1.5 and 3.5, and rarely exceed 4.022. In this study, the Shannon-Weiner index, H, of the microbial community in the tar sands was 3.072 and the Shannon evenness, E, was 1.009. These values indicate a more diverse microbial community and an even distribution of the species within the tar sands.
This study investigated the indigenous microbial communities of the tar sands of Utah in an effort to understand the structure and the functional diversity of the microbial community. The substrate utilization patterns of resident microbial communities and the identification of the hydrocarbonoclastic microorganisms in the tar sands provide valuable baseline information than can be used for hydrocarbon bioremediation and for devising biotechnological approaches to tar sands bioremediation efforts.
Dataset 1. Raw data obtained from the community-level physiological profile, 10.5256/f1000research.16126.d21890923.
The DNA sequences from this metagenomic project are available from the Sequence Read Archive/European Nucleotide Archive, accession number SRR1699470.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)