Faasse S, Braun H and Vos M. The role of NAFLD in cardiometabolic disease: an update [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):170 (https://doi.org/10.12688/f1000research.12028.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
Miriam Vos
Roles:
Conceptualization,
Visualization,
Writing – Original Draft Preparation,
Writing – Review & Editing
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease in the world, yet the complex pathogenesis remains to be fully elucidated. The prevalence of NAFLD has risen precipitously in recent years and is now a leading indication for liver transplantation. New waitlist registrants with non-alcoholic steatohepatitis–induced cirrhosis increased by 170% from 2004 to 2013. In addition, patients with NAFLD are at increased risk of both cardiovascular disease and type II diabetes. In this update, recent studies contributing to the understanding of the place of NAFLD in cardiometabolic disease will be discussed.
Keywords
non-alcoholic fatty liver disease, cardiometabolic disease, type 2 diabetes mellitus
Corresponding author:
Miriam Vos
Competing interests:
SF and HB declare that they have no competing interests. MBV has NAFLD-related research support or in-kind research services from Resonance Health, Nutrition Science Initiative, AMRA, Siemens, Shire, Target PharmaSolutions, Labcorp, and Perspectum and serves as an NAFLD consultant for Allergan, AMRA, Boehringer Ingelheim, Bristol-Myers Squibb, Immuron, Intercept, Shire, and Target PharmaSolutions.
Grant information:
This work was supported through a grant provided by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) R21 HD0809056-01.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Non-alcoholic fatty liver disease (NAFLD), now the most common liver disease in the world, is defined as the accumulation of fat in the liver beyond 5% of total liver weight in the absence of excessive alcohol use and other pathogenic sources such as viral hepatitis, Wilson disease, alpha-1-antitrypsin deficiency, autoimmune hepatitis, or other metabolic diseases1–4. The estimated prevalence of NAFLD in the US is now 19%, and worldwide prevalence estimates are between 25.24 and 32.5%5,6. The marked increase in NAFLD prevalence has occurred concomitantly with increases in rates of obesity and the metabolic syndrome (MS) worldwide. Obesity itself is heterogenic, and a greater understanding of the relevance of body fat distribution patterns to NAFLD has developed recently as a growing number of studies using magnetic resonance imaging (MRI) to measure fat distribution have been published7–9. The previous mainstay was dual-energy x-ray absorptiometry, useful for visceral fat10, muscle mass, and total adiposity, but it misses hepatic fat. Body mass index (BMI) can both underestimate disease, as in the case of metabolically obese, normal-weight individuals11, and overestimate disease, as in the case of obese (BMI of more than 30 kg/m2) but metabolically healthy people.
People in the metabolically unhealthy category, regardless of BMI, tend to have increased visceral adipose tissue (VAT)12. VAT is metabolically active and has been thought to be complicit in the dysmetabolic syndrome13. Individuals with a higher proportion of VAT are more likely to have NAFLD14 and a higher risk of associated dysmetabolic disease15,16. This leads to questions of which drives which: hepatic fat leading to VAT, or vice versa? There are two studies that demonstrate in both adults and adolescents that intrahepatic fat, rather than VAT, is most closely associated with insulin resistance (IR), suggesting that intrahepatic fat is the primary driver of metabolic dysfunction in the centrally located adiposity phenotype7,8. Within the liver, de novo lipogenesis (DNL) is the primary dysfunction of lipid metabolism leading to steatosis accumulation. In healthy metabolism, DNL is not a major contributor of lipids in the liver. However, in NAFLD, DNL is abnormally upregulated17, and it increases further in the setting of weight gain18. In NAFLD, approximately one fourth of the fat in the liver is directly from DNL19.
Amino acid metabolism is another important area of disturbance in NAFLD, and newer techniques such as high-throughput, high-resolution metabolomics have advanced studies in this area. Specifically, levels of branched chain amino acids (leucine, isoleucine, and valine) and aromatic amino acids (tyrosine) have been found to be altered in NAFLD20,21. Notably, a similar amino acid profile was also found to predict risk of future type 2 diabetes mellitus (T2DM)22. In pediatric patients with NAFLD, tyrosine metabolism was found to be most significantly altered in adolescent patients, and a recent study shows that elevated levels of tyrosine, glutamate, and glutamate/(serine+glycine) ratio (that is, GSG index) were associated with IR and that alanine, glutamate, isoleucine, and valine were increased in non-obese patients with NAFLD21,23. More studies are needed to understand whether dysregulated amino acids represent promising biomarkers or future therapeutic targets for NAFLD.
Genetic factors
As is typical of many chronic diseases, NAFLD is a genetic condition with an environmental trigger that induces the expression of the phenotype. In the case of NAFLD, this trigger appears to be, at a minimum, a hypercaloric condition. The most established genotype complicit in liver steatosis is a mutation of the rs738409 allele of patatin-like phospholipase domain (PNPLA3)24. The mutation is a single-nucleotide polymorphism (SNP) of codon 148, resulting in a missense mutation of isoleucine to methionine (I148M)25. The homozygous variant is most commonly found in Hispanics. A recent article found that adiposity potentiates the effect of this missense mutation, resulting in a higher risk for development of NAFLD as BMI increases in patients with this particular variant26. Interestingly, another variant allele of PNPLA3 (rs6006460) that results in a serine-to-isoleucine (S453I) missense appears to have a hepatoprotective effect, associated with lower intrahepatic fat content, and is most commonly found in African-Americans, an ethnic group that has a relatively low prevalence of NAFLD27. Other SNPs have been identified in the pathogenesis of NAFLD, glucokinase regulatory protein (GCKR) rs126032628, and transmembrane 6 superfamily member 2 (TM6SF2) rs5854292629, and when assessed with PNPLA3 rs6006460, these three SNPs seem to have an additive effect on determining intrahepatic fat content29.
Prenatal exposure
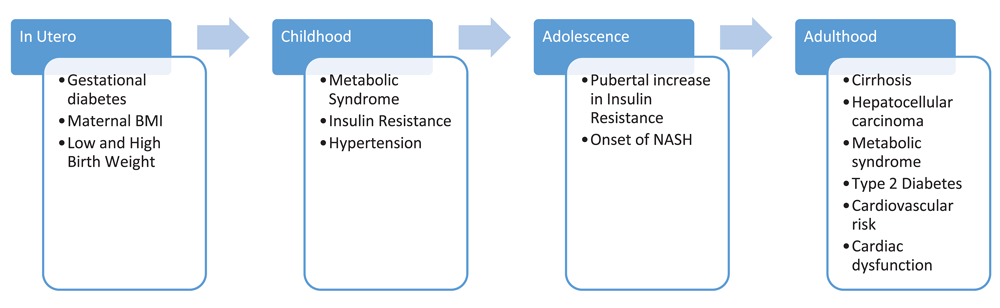
It is vital to consider NAFLD within the life span of cardiometabolic disease as it is not an isolated disease (Figure 1)30. Some important studies have emerged that suggest prenatal origins for NAFLD. Friedman et al. conducted an elegant set of experiments in rhesus monkeys and demonstrated that a high-fat diet in insulin-resistant pregnant monkeys predicted fatty liver in the offspring, regardless of the diet (healthy or not) given to the infant monkeys31,32. Limited but intriguing evidence in humans supports this finding. Two recent MRI-based studies found that higher maternal BMI positively correlates with both higher postnatal infant adipose tissue and intrahepatic fat content, suggesting that the burden of intrahepatic fat begins in utero33,34. Similarly, infants born to obese mothers with gestational diabetes (GDM) had, on average, 68% more hepatic fat content by MRI quantification than those born to normal-weight women34. An autopsy study of stillborn babies showed that histopathologic hepatic steatosis was significantly more prevalent in children whose mothers had maternal GDM (79%) compared with controls (17%) (P <0.001)35. Together, these studies suggest that maternal IR may program the liver during development and drive hepatocyte function toward steatosis. More information is needed regarding maternal environmental exposures and the effect on NAFLD predisposition in children.
Figure 1. Temporality of risk factors, comorbidities, and potential consequences of non-alcoholic fatty liver disease.
These findings suggest a common exposure or complementary or additive relationship between non-alcoholic fatty liver disease and other metabolic diseases. BMI, body mass index; NASH, non-alcoholic steatohepatitis.
Non-alcoholic fatty liver disease in childhood and adolescence
Moving forward to early childhood, there are currently little data on the status of the liver in children who are one to seven years old, but markers of MS can be seen in preschool-aged children. Prenatal and perinatal exposures of GDM and high birth weight are both established risk factors for MS in childhood36. In addition, both high and low birth weight are correlated not only with NAFLD but also with increased risk of more advanced liver disease37. Some evidence for NAFLD in preschool-aged children within high risk groups also exists. In a study mostly of Hispanic children in an obesity clinic, about one fourth of children starting at preschool age had elevated alanine aminotransferase (ALT) for age, and this proportion rose to nearly half by middle-school age38.
NAFLD in most children is diagnosed in the peripubertal period. During puberty, IR transiently increases39,40, and children who are obese or have existing IR have a greater increase in IR during puberty, which may contribute to greater pathology in the liver41. Histologically, children in puberty can display greater hepatic steatosis and more severe portal inflammation than adults, whereas lobular inflammation and ballooning appear milder in children compared with adults42.
Non-alcoholic fatty liver disease as a risk factor of cardiometabolic disease
The increase in IR and worsening of NAFLD can be somewhat of a “chicken or the egg” question. Few longitudinal studies exist in children to help answer this, but in a study of Japanese adults, after adjustment for other confounding factors, patients with fatty liver were more likely (hazard ratio (HR) 1.49) to become dysmetabolic compared with their counterparts without fatty liver43. This supports the view of NAFLD as a driver of ongoing IR, visceral adiposity, and metabolic dysfunction15,44.
There is convincing evidence that NAFLD is an independent risk factor for cardiovascular disease events even after adjustment for conventional risk factors (odds ratio 4.12, 95% confidence interval (CI) 1.58 to 10.75, P = 0.004)45. Significantly more patients with NAFLD had either coronary artery disease (7.5% versus 1.4%) or stroke (0.9% versus 0.2%) than non-NAFLD control patients46. Data suggest that cardiac intima-media thickness (CIMT), an early predictor of atherosclerosis, and Framingham risk score significantly increase across quartiles of the fatty liver index (an accurate predictor of hepatic teatosis on ultrasonography)47,48. In addition, patients who developed hepatic steatosis showed an increase in CIMT whereas those without did not see a significant change47. In regard to cardiac dysfunction risk, patients with NAFLD, particularly those with non-alcoholic steatohepatitis, have worsened right ventricular function compared with non-NAFLD counterparts49. Coupled with type 2 diabetes, the risk for left ventricular dysfunction is also higher: patients with both T2DM and NAFLD have greater left ventricular diastolic dysfunction than those with T2DM alone50.
There is also evidence that NAFLD may increase risk of T2DM44,51. One study found the prevalence of T2DM to be higher in patients with NAFLD when compared with their obese, non-NAFLD counterparts (6.5% versus <1%). Notably, girls with NAFLD were more likely to have T2DM than boys with NAFLD, despite an overall higher prevalence of NAFLD in boys52. A longitudinal cohort study found that patients with only MS had a significantly higher risk of developing T2DM than those with NAFLD and no MS but that patients with both NAFLD and MS had the greatest risk of developing T2DM, almost four times greater than those with only NAFLD and two times greater than those with only MS. In addition, among those with MS, NAFLD significantly increased risk of T2DM (HR 1.93, 95% CI 1.38–2.71)53. These authors hypothesized that the absence of an independent association between NAFLD and T2DM in those patients without MS may be due to differences in the severity of NAFLD53. This theory is further supported by an independent study that found that NAFLD severity was associated with increased T2DM incidence even after controlling for MS and other predictors of T2DM54.
Conclusions
In summary, the intertwined stories of NAFLD and cardiometabolic dysregulation appear far more complex than previously hypothesized. In light of recent evidence suggesting that NAFLD may develop in utero and that NAFLD increases risk of both cardiovascular disease and T2DM, we contend that it is important to assess and treat NAFLD in childhood in order to prevent progression to these diseases. It is no longer possible to view NAFLD solely as a liver disease resulting from metabolic dysregulation; rather, it may be a causative factor in the spiral toward T2DM and cardiovascular events. The typical NAFLD clinical assessment is currently confined to liver pathology (steatosis, fibrosis, and screening for consequential cirrhosis and hepatocellular carcinoma), but this growing body of evidence emphasizes the importance of a broader patient assessment and demonstrates that NAFLD is a disease with systemic pathophysiologic consequences.
The practical applications of these conclusions are several. More knowledge is needed regarding hepatic fat in infants and young children and to understand whether hepatic steatosis disappears and reappears or is sustained from birth. This will require the development of a better and safer non-invasive measurement of fat in very young children. Furthermore, children in groups known to be at high risk for NAFLD should be studied longitudinally so that the onset of the liver disease can be better understood. This will be critical for developing a public health approach to prevent NAFLD and to test interventions that interrupt its course at the onset. Lastly, emerging evidence now suggests that, once children develop NAFLD, they are at high risk for the development of T2DM. This needs to be confirmed in a large-scale, long-term longitudinal study of pediatric NAFLD, and if it is confirmed, this population could be given priority for T2DM prevention programs.
Competing interests
SF and HB declare that they have no competing interests. MBV has NAFLD-related research support or in-kind research services from Resonance Health, Nutrition Science Initiative, AMRA, Siemens, Shire, Target PharmaSolutions, Labcorp, and Perspectum and serves as an NAFLD consultant for Allergan, AMRA, Boehringer Ingelheim, Bristol-Myers Squibb, Immuron, Intercept, Shire, and Target PharmaSolutions.
Grant information
This work was supported through a grant provided by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) R21 HD0809056-01.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Faculty Opinions recommended
References
1.
Schwimmer JB, Deutsch R, Kahen T, et al.:
Prevalence of fatty liver in children and adolescents.
Pediatrics.
2006; 118(4): 1388–93. PubMed Abstract
| Publisher Full Text
2.
Welsh JA, Karpen S, Vos MB:
Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010.
J Pediatr.
2013; 162(3): 496–500.e1. PubMed Abstract
| Publisher Full Text
| Free Full Text
3.
Wong RJ, Aguilar M, Cheung R, et al.:
Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States.
Gastroenterology.
2015; 148(3): 547–55. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
5.
Lazo M, Hernaez R, Eberhardt MS, et al.:
Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994.
Am J Epidemiol.
2013; 178(1): 38–45. PubMed Abstract
| Publisher Full Text
| Free Full Text
6.
Younossi ZM, Koenig AB, Abdelatif D, et al.:
Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes.
Hepatology.
2016; 64(1): 73–84. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
7.
D'Adamo E, Cali AM, Weiss R, et al.:
Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents.
Diabetes Care.
2010; 33(8): 1817–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
10.
Kaul S, Rothney MP, Peters DM, et al.:
Dual-energy X-ray absorptiometry for quantification of visceral fat.
Obesity (Silver Spring).
2012; 20(6): 1313–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
11.
Ruderman NB, Schneider SH, Berchtold P:
The "metabolically-obese," normal-weight individual.
Am J Clin Nutr.
1981; 34(8): 1617–21. PubMed Abstract
| Publisher Full Text
12.
Neeland IJ, Ayers CR, Rohatgi AK, et al.:
Associations of visceral and abdominal subcutaneous adipose tissue with markers of cardiac and metabolic risk in obese adults.
Obesity (Silver Spring).
2013; 21(9): E439–47. PubMed Abstract
| Publisher Full Text
| Free Full Text
13.
Fontana L, Eagon JC, Trujillo ME, et al.:
Visceral fat adipokine secretion is associated with systemic inflammation in obese humans.
Diabetes.
2007; 56(4): 1010–3. PubMed Abstract
| Publisher Full Text
17.
Lambert JE, Ramos-Roman MA, Browning JD, et al.:
Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease.
Gastroenterology.
2014; 146(3): 726–35. PubMed Abstract
| Publisher Full Text
19.
Donnelly KL, Smith CI, Schwarzenberg SJ, et al.:
Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease.
J Clin Invest.
2005; 115(5): 1343–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
21.
Jin R, Banton S, Tran VT, et al.:
Amino Acid Metabolism is Altered in Adolescents with Nonalcoholic Fatty Liver Disease-An Untargeted, High Resolution Metabolomics Study.
J Pediatr.
2016; 172: 14–19.e5. PubMed Abstract
| Publisher Full Text
| Free Full Text
24.
Arslanow A, Stokes CS, Weber SN, et al.:
The common PNPLA3 variant p.I148M is associated with liver fat contents as quantified by controlled attenuation parameter (CAP).
Liver Int.
2016; 36(3): 418–26. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
25.
Liu W, Baker RD, Bhatia T, et al.:
Pathogenesis of nonalcoholic steatohepatitis.
Cell Mol Life Sci.
2016; 73(10): 1969–87. PubMed Abstract
| Publisher Full Text
28.
Santoro N, Zhang CK, Zhao H, et al.:
Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents.
Hepatology.
2012; 55(3): 781–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
30.
Cioffi CE, Welsh JA, Cleeton RL, et al.:
Natural History of NAFLD Diagnosed in Childhood: A Single-Center Study.
Children (Basel).
2017; 4(5): pii: E34. PubMed Abstract
| Publisher Full Text
| Free Full Text
31.
Thorn SR, Baquero KC, Newsom SA, et al.:
Early life exposure to maternal insulin resistance has persistent effects on hepatic NAFLD in juvenile nonhuman primates.
Diabetes.
2014; 63(8): 2702–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
33.
Modi N, Murgasova D, Ruager-Martin R, et al.:
The influence of maternal body mass index on infant adiposity and hepatic lipid content.
Pediatr Res.
2011; 70(3): 287–91. PubMed Abstract
| Publisher Full Text
34.
Brumbaugh DE, Tearse P, Cree-Green M, et al.:
Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes.
J Pediatr.
2013; 162(5): 930–6.e1. PubMed Abstract
| Publisher Full Text
| Free Full Text
36.
Boney CM, Verma A, Tucker R, et al.:
Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus.
Pediatrics.
2005; 115(3): e290–6. PubMed Abstract
| Publisher Full Text
38.
Beacher DR, Ariza AJ, Fishbein MH, et al.:
Screening for elevated risk of liver disease in preschool children (aged 2-5 years) being seen for obesity management.
SAGE Open Med.
2014; 2: 2050312114555211. PubMed Abstract
| Publisher Full Text
| Free Full Text
39.
Guzzaloni G, Grugni G, Mazzilli G, et al.:
Comparison between beta-cell function and insulin resistance indexes in prepubertal and pubertal obese children.
Metabolism.
2002; 51(8): 1011–6. PubMed Abstract
| Publisher Full Text
40.
Moran A, Jacobs DR Jr, Steinberger J, et al.:
Insulin resistance during puberty: results from clamp studies in 357 children.
Diabetes.
1999; 48(10): 2039–44. PubMed Abstract
| Publisher Full Text
42.
Takahashi Y, Inui A, Fujisawa T, et al.:
Histopathological characteristics of non-alcoholic fatty liver disease in children: Comparison with adult cases.
Hepatol Res.
2011; 41(11): 1066–74. PubMed Abstract
| Publisher Full Text
43.
Hashimoto Y, Hamaguchi M, Fukuda T, et al.:
Fatty liver as a risk factor for progression from metabolically healthy to metabolically abnormal in non-overweight individuals.
Endocrine.
2017; 57(1): 89–97. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
45.
Hamaguchi M, Kojima T, Takeda N, et al.:
Nonalcoholic fatty liver disease is a novel predictor of cardiovascular disease.
World J Gastroenterol.
2007; 13(10): 1579–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
46.
Mahfood Haddad T, Hamdeh S, Kanmanthareddy A, et al.:
Nonalcoholic fatty liver disease and the risk of clinical cardiovascular events: A systematic review and meta-analysis.
Diabetes Metab Syndr.
2017; 11 Suppl 1: S209–S216. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
49.
Sunbul M, Kivrak T, Durmus E, et al.:
Nonalcoholic Steatohepatitis Score is an Independent Predictor of Right Ventricular Dysfunction in Patients with Nonalcoholic Fatty Liver Disease.
Cardiovasc Ther.
2015; 33(5): 294–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
53.
Bae JC, Kim SK, Han JM, et al.:
Additive effect of non-alcoholic fatty liver disease on the development of diabetes in individuals with metabolic syndrome.
Diabetes Res Clin Pract.
2017; 129: 136–43. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
54.
Park SK, Seo MH, Shin HC, et al.:
Clinical availability of nonalcoholic fatty liver disease as an early predictor of type 2 diabetes mellitus in Korean men: 5-year prospective cohort study.
Hepatology.
2013; 57(4): 1378–83. PubMed Abstract
| Publisher Full Text
SF and HB declare that they have no competing interests. MBV has NAFLD-related research support or in-kind research services from Resonance Health, Nutrition Science Initiative, AMRA, Siemens, Shire, Target PharmaSolutions, Labcorp, and Perspectum and serves as an NAFLD consultant for Allergan, AMRA, Boehringer Ingelheim, Bristol-Myers Squibb, Immuron, Intercept, Shire, and Target PharmaSolutions.
This work was supported through a grant provided by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) R21 HD0809056-01.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Faasse S, Braun H and Vos M. The role of NAFLD in cardiometabolic disease: an update [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):170 (https://doi.org/10.12688/f1000research.12028.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Santoro N. Reviewer Report For: The role of NAFLD in cardiometabolic disease: an update [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):170 (https://doi.org/10.5256/f1000research.13013.r30519)
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Santoro N. Reviewer Report For: The role of NAFLD in cardiometabolic disease: an update [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):170 (https://doi.org/10.5256/f1000research.13013.r30519)
Targher G. Reviewer Report For: The role of NAFLD in cardiometabolic disease: an update [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):170 (https://doi.org/10.5256/f1000research.13013.r30518)
NOTE: it is important to ensure the information in square brackets after the title is included in this citation.
Reviewer Report09 Feb 2018
Giovanni Targher,
Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, University and Azienda Ospedaliera Universitaria Integrata of Verona, Verona, Italy
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Targher G. Reviewer Report For: The role of NAFLD in cardiometabolic disease: an update [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):170 (https://doi.org/10.5256/f1000research.13013.r30518)
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)