Keywords
environmental DNA, data visualization, citizen science, community science, shiny, metabarcoding, education, community ecology
This article is included in the Ecology and Global Change gateway.
This article is included in the RPackage gateway.
This article is included in the Interactive Figures collection.
environmental DNA, data visualization, citizen science, community science, shiny, metabarcoding, education, community ecology
The targeted amplification and sequencing of DNA that living organisms shed into their physical environment, termed “environmental DNA (eDNA) metabarcoding,” is revolutionizing microbiology, ecology, and conservation research (Deiner et al., 2017; Taberlet et al. 2012). Sequencing of eDNA extracted from field-collected soil, water, or sediment samples can yield insight into a range of questions, from profiling the composition of ancient plant and animal communities (Pedersen et al., 2015), to monitoring populations of rare or endangered species (Balasingham et al., 2018). As the cost of eDNA metabarcoding declines and sample collection techniques become more streamlined (e.g. Thomas et al. (2018)), professional research scientists are increasingly using eDNA metabarcoding as a platform to engage a diversity of community partners, including natural resource managers, undergraduate students, and citizen scientists in primary research. However, developing robust and impactful community science programs that engage community partners in all steps of the research process remains a challenge.
eDNA metabarcoding-based projects work well for programs that partner researchers with community scientists because non-experts can be quickly trained to collect samples in the field, and because eDNA metabarcoding is an exciting framework for research pertinent to disciplines such as medicine, agriculture, ecology, and geography (Deiner et al., 2017). Community partners in such programs can have heterogeneous backgrounds, ranging from curious members of the public for whom collecting samples in the field is their first scientific research experience (e.g. University of California’s CALeDNA program), to professional natural resource managers who regularly collaborate with research scientists (e.g. Center for Ocean Solutions’ eDNA project). A key ingredient to promote sustained success of such programs is that community partners should be able to engage across multiple stages of the research project, not only in sample collection (European Citizen Science Association, 2015; Pandya, 2012). This can be a challenge for community science programs because although it is relatively easy to train community partners to collect eDNA samples, it is far more challenging to train them to independently visualize and analyze results from these studies. Indeed, learning the bioinformatic tools necessary for managing the large, multidimensional datasets generated in these studies can be difficult for professional researchers (Carey & Papin, 2018), let alone for the non-technical audience of some community science programs.
To address this challenge, we created the R package “ranacapa”, at the core of which is a Shiny web app that can be used to visualize results from eDNA sequencing studies and perform simple community ecology analyses. ranacapa complements existing visualization platforms (e.g. Phinch (Bik & Phinch Interactive, 2014), Phyloseq-Shiny (McMurdie & Holmes, 2015), QIIME2 Viewer), because in addition to interactive visualizations, ranacapa includes brief explanations of several core analyses used in eDNA studies and includes links to additional educational resources. ranacapa works with community matrices generated via QIIME (Caporaso et al., 2010) or the Anacapa sequence analysis pipeline, the latter being used extensively by the CALeDNA program.
Here, we describe the package and how it is used by two community science partnerships based at the University of California, Los Angeles (UCLA): first, a collaboration between eDNA researchers and resource managers at the National Park Service, and second, a partnership between community ecology researchers and an undergraduate microbiology course at UCLA. As we show in the Use cases, empowering community partners to interact with the data and perform simple but insightful community ecology analyses can help make these collaborations more enriching and valuable to both parties.
At the core of ranacapa is a Shiny web app (Chang et al., 2018), which is available at http://gauravsk.shinyapps.io/ranacapa or with ranacapa::runRanacapa(). The package also includes two categories of helper functions (Table 1) that transform user-uploaded taxonomy and metadata tables into R objects that can be visualized and analyzed using the Phyloseq (McMurdie & Holmes, 2013) and Vegan (Oksanen et al., 2018) packages. ranacapa is available for installation from Github or CRAN:
devtools::install_github("gauravsk/ranacapa") install.packages("ranacapa")
| Name | Description |
|---|---|
| scrub_seqNum_column | Removes any "xxx_seq_number" columns from the input taxonomy file if present (depends on which version of Anacapa was used to assign taxonomy) |
| scrub_taxon_paths | Replaces empty cells in input taxonomy tables with “Unknown” |
| validate_input_files | Verifies that the input taxonomy file and input mapping file meet specifications |
| convert_biom_to_taxon_table | Converts a phyloseq-imported biom table into an Anacapa-formatted taxonomy table |
| group_anacapa_by_taxonomy | Summarizes a site-abundance table from the Anacapa pipeline to each unique taxon |
| categorize_continuous_vector | Categorizes a continuous vector into low, medium, and high |
| convert_anacapa_to_phyloseq | Converts a site-abundance table from the Anacapa pipeline and the associated metadata file into a phyloseq object |
| vegan_otu | Creates a community matrix in the vegan package style using a phyloseq object and an otu_table object |
| custom_rarefaction | Rarefies a phyloseq object to a custom sample depth and with a given number of replicates |
| pairwise_adonis1 | Wrapper function for multilevel pairwise comparison |
| ggrare2 | Makes a rarefaction curve using ggplot2 |
| runRanacapaApp | Runs the ranacapa Shiny app with tabs for interactive visualizations and statistical analyses |
1 adopted from https://github.com/pmartinezarbizu/pairwiseAdonis (GPL-3 License)
2 adopted from https://github.com/mahendra-mariadassou/phyloseq-extended (GPL-3 License)
The ranacapa Shiny app allows users to interact with eDNA results through statistical summaries and interactive plots, displayed in the following tabs:
• Sequencing depth: Introduces the potential for variation in sequencing depth among samples and explains the basic logic behind rarefying samples in metagenomics studies (Figure 1). Users can rarefy the dataset to a sampling depth, or can proceed through the rest of the app without rarefying samples. The documentation acknowledges recent disagreement regarding the value of rarefying in metabarcoding and eDNA sequencing studies (McMurdie & Holmes, 2014).
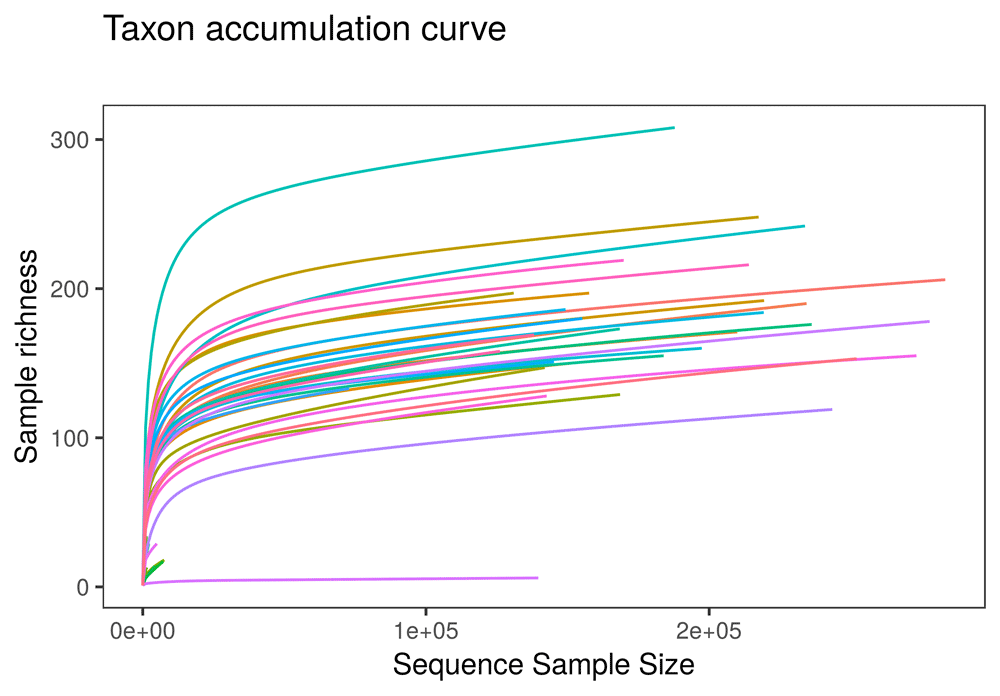
The online version of this figure is interactive.
• Taxonomy heatmap: Shows the taxon-by-sample matrix as an interactive heatmap made using heatmaply::heatmaply() (Galili et al., 2018), where the color of each cell represents the number of times a given taxon was sequenced in a sample (Figure 2). Users can filter the taxon list by selecting or deselecting specific taxa.
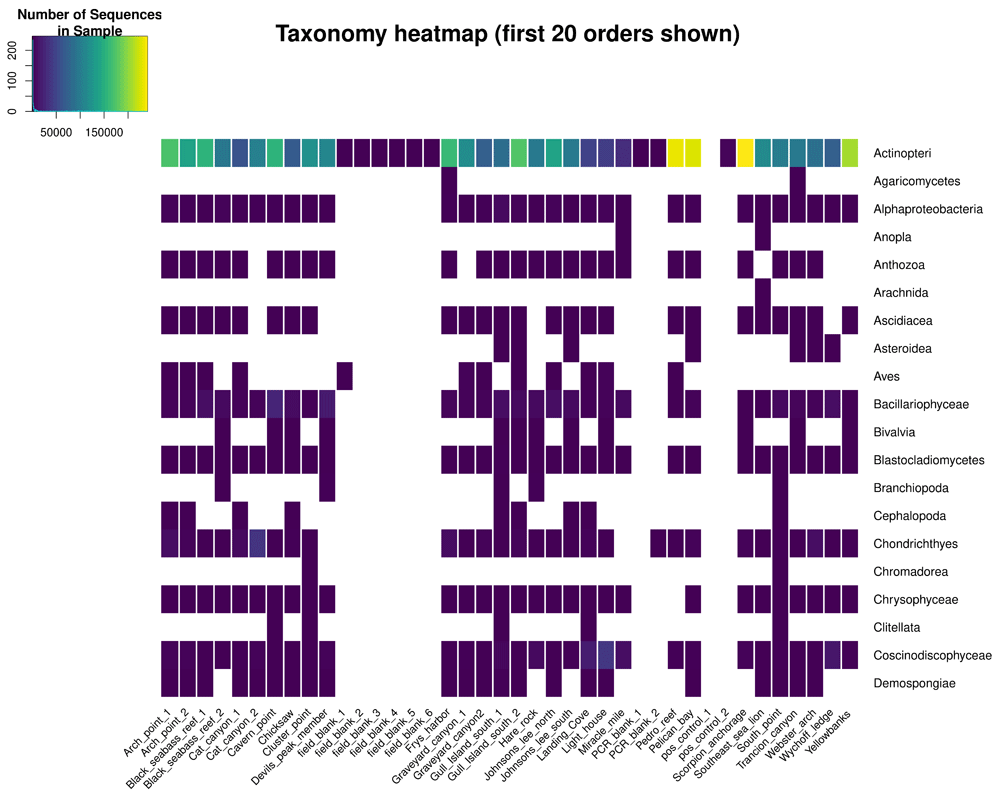
Taxonomy is shown at the Order level in this figure; in the app, users can choose the taxonomic level to show in the heatmap. Users can also select or deselect individual taxa to be shown in the heatmap. The online version of this figure is interactive.
• Taxonomy barplot: Shows the taxonomy-by-sample matrix as an interactive barplot (Figure 3).
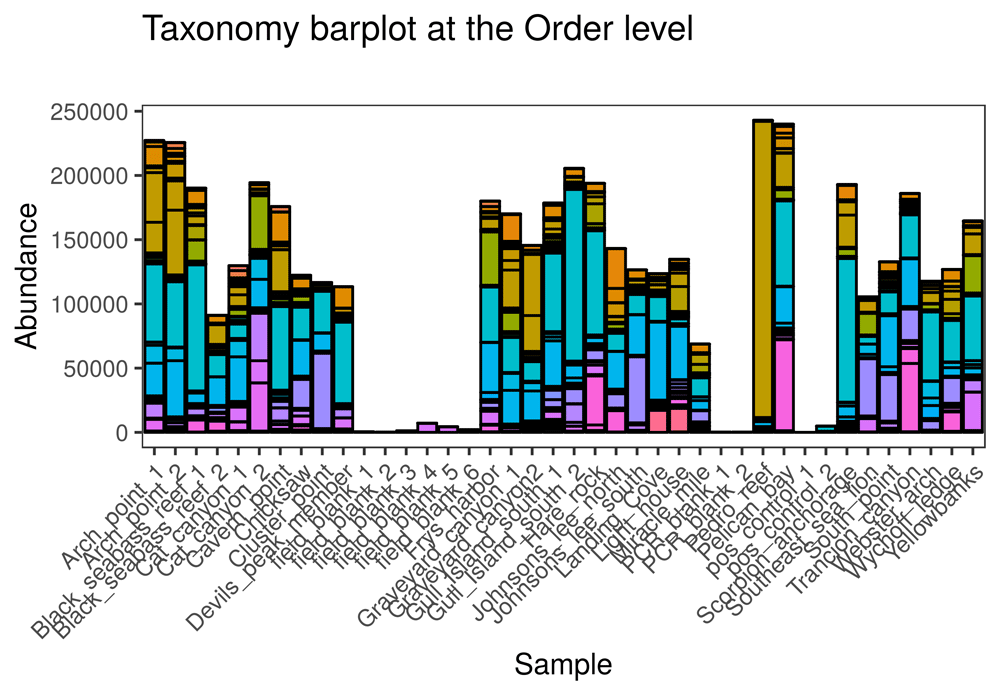
Taxonomy is shown at the Order level in this figure; in the app, users can choose the taxonomic level to show in the barplot. The online version of this figure is interactive.
• Alpha diversity plots: Introduces the concept of alpha diversity as the local diversity measured in a single habitat or sample. Users can plot alpha diversity as observed taxon richness or as Shannon diversity per sample, or can group samples according to a variable in the metadata file (Figure 4).
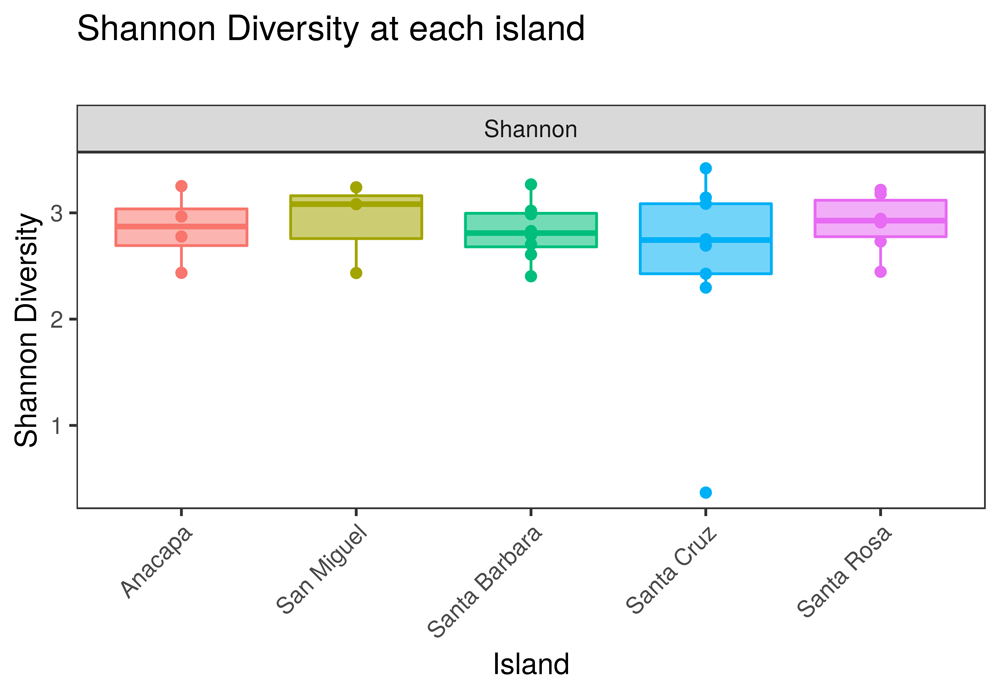
Users can select the X-axis variable using a dropdown menu in the app. The online version of this figure is interactive.
• Alpha diversity statistics: Allows users to choose a variable from the metadata, and generates an alpha diversity ANOVA table according to the user-selected variable. The tab also shows the output from a post-hoc Tukey test.
• Beta diversity plots: Introduces the concept of beta diversity as the turnover in species composition across habitats (or samples). The tab includes an ordination plot generated by phyloseq::plot_ordination(), which in turn uses an ordination object made with phyloseq::ordinate(., method = "PCoA"). Points on the PCoA plot are colored according to a user-selected metadata variable (Figure 5).
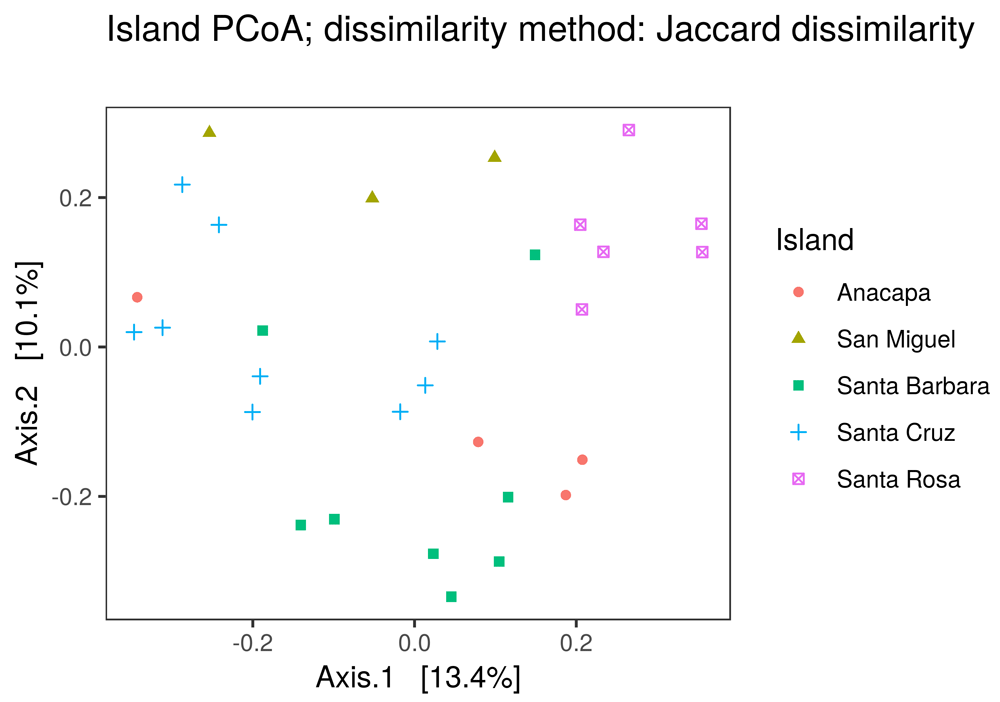
Users can select the grouping variable with a dropdown menu in the app. The online version of this figure is interactive.
The beta diversity plots tab also includes a dendrogram that groups sites based on Ward’s cluster analysis (stats::hclust(distance_object, method = "ward.d2")), where distance_object is made using phyloseq::distance(). For both figures, users can toggle between using Jaccard and Bray-Curtis dissimilarity.
• Beta diversity statistics: Shows results from two statistical tests of species turnover across sites. The first test is a multivariate ANOVA of taxon turnover across sites, implemented with vegan::adonis(). The second statistical test, which is implemented with vegan::betadisper(), is of heterogeneity of variances among samples. This test compares the degree of sample-to-sample variation within habitats (or within other user-selected groups).
ranacapa depends on Bioconductor v 3.7, which in turn relies on R v 3.5.0. The Shiny app has been tested on Chrome and Firefox on Windows, Mac-OSX, and Ubuntu.
The ranacapa Shiny app requires two input files. The first requirement is a taxon-by-sample matrix, uploaded either as a rich, dense .biom table, or as a tab-separated .txt file. Qiime2-generated .qza files generated by QIIME2 are not immediately suitable for ranacapa, as they do not contain full taxonomy information. If the site-by-species matrix is uploaded as a .txt file, the file should match the specifications of the output files from the Anacapa eDNA sequence analysis pipeline. In Anacapa output, each row represents a taxonomic identification, and each column (save one) represents the number of times that taxon appears in each sequenced sample. One column, named sum.taxonomy must contain the taxonomic identification, with taxonomic rank separated by a semicolon, e.g. “Chordata;Actinopteri;Chaetodontiformes;Chaetodontidae;Chaetodon;Chaetodon reticulatus .” A valid input file is structured as follows:
sum.taxonomy Arch_point_1 Arch_point_2 Black_seabass_reef_1 Black_seabass_reef_2
<full path> 0 0 0 0
<full path> 0 0 43 87
<full path> 0 0 0 0
<full path> 0 0 0 0
<full path> 24 36 30 16
<full path> 0 0 0 0
<full path> 0 0 0 0
<full path> 0 0 16 177
<full path> 0 0 0 0
<full path> 0 0 0 0
The second requirement is a tab-separated .txt file that contains sample metadata. The first column in the metadata file should match the sample names in the taxonomy table; the remaining columns contain sample information for each of the samples in the taxon-by-site matrix. The metadata should contain categorical variables with two or more categories per variable. A valid metadata file for the taxonomy table above is structured as follows:
Sample Sample_or_Control Island Protection Locality
Black_seabass_reef_1 Sample Anacapa MPA Black_seabass_reef
Arch_point_1 Sample Santa Barbara non-MPA Arch_point
Arch_point_2 Sample Santa Barbara non-MPA Arch_point
Black_seabass_reef_2 Sample Anacapa MPA Black_seabass_reefThe ranacapa function validate_input_files() verifies that both the taxonomy table and the metadata files match structural requirements, which are documented in the function help files.
We expect that researchers with expertise in bioinformatics will use the sequence analysis pipeline of their choice to assign taxonomy to eDNA datasets, and generate clean taxonomy and metadata files that can be visualized in ranacapa. Researchers can share these files with their partners, and emphasize the analyses or visualizations most appropriate to their use case. We now show how ranacapa can facilitate authentic communication between researchers and community partners in two settings.
A team of UCLA researchers partnered with resource managers at the Channel Islands National Park Service to assess the potential for eDNA as a biodiversity monitoring tool to supplement time-intensive visual biodiversity surveys in the Southern California Channel Islands (Deiner et al., 2017; Lessios, 1996; Usseglio, 2015). For this partnership, resource managers collected and filtered 30 unique one-liter water samples for eDNA analysis at permanent monitoring sites inside and adjacent to protected areas, and research scientists at UCLA performed eDNA sequencing of the mitochondrial 12S (Miya et al., 2015) and CO1 (Leray et al., 2013) genes, targeting bony fishes, elasmobranches, and invertebrate taxa. The researchers processed sequences and assigned taxonomy using the Anacapa pipeline, and shared results with the resource managers using the ranacapa Shiny app.
The taxonomy heatmap of species detected using the 12S and CO1 metabarcodes (Figure 2) was the most valuable visualization to this collaboration, because it allowed the resource managers to filter the large observed species list down to a particular set of key taxa that they regularly monitor. The heatmap showed that this pilot study detected 36 of the 70 key metazoans at the species level, and the remaining 34 at the genus, family, or order level. This indicates that eDNA-based studies can provide critical information for ongoing management efforts and provide new insights into the spatial and temporal distributions of these species. The value of ranacapa in this scenario was to quickly sort through long species lists generated by eDNA sequencing to highlight the strengths and weaknesses in using eDNA to monitor diversity in the Channel Islands. The data from this study are packaged as the demo dataset for the ranacapa Shiny app and are available online (Kandlikar et al., 2018a).
A team of community ecology and environmental DNA researchers in the CALeDNA program collaborated with instructors of a research-based environmental microbiology course at UCLA (Shapiro et al., 2015), in which students used eDNA metabarcoding to study the impact of a local wildfire on the plant and soil microbial community. The goal of this twenty-week course was to provide undergraduate students an authentic experience in basic microbiology and microbial community ecology research. Over the first ten weeks, eDNA researchers on the instructional team sequenced the ITS2 (Gu et al., 2013) and 16S SSU RNA (Caporaso et al., 2012) metabarcoding regions from student-collected soil samples and used the Anacapa pipeline to generate taxon-by-sample tables.
The course instructors used the ranacapa Shiny app to introduce students to the structure of eDNA sequencing results. The students were encouraged to explore data and perform the statistical analyses most pertinent to the hypotheses they had formed at the beginning of the course. A key benefit of using ranacapa was that despite having no prior bioinformatics experience, students could begin exploring the biodiversity in their samples in a matter of minutes by using the online instance of the Shiny app. This allowed the instructors to focus classroom time on biological questions rather than on troubleshooting bioinformatics problems, as had been the case in previous sessions of the course. The course instructors noted that visualizing eDNA data in ranacapa helped students understand the relationships between taxon-by-site matrices and the various metadata they had collected in the field. By significantly reducing the time and difficulty in visualizing basic biodiversity patterns, ranacapa helped students develop and pursue more sophisticated analyses during the remainder of the course, using tools such as STAMP (Parks et al., 2014) and PICRUSt (Langille et al., 2013). The taxonomy tables and metadata files used in this course are available online (Kandlikar et al., 2018b).
Metabarcode sequencing of environmental DNA is becoming a key tool in a wide variety of ecological studies, and results from these studies are of interest to a broad audience. Our R package and Shiny app ranacapa helps users conduct exploratory analyses and visualizations on eDNA datasets, and is a step toward more fully engaging participants in all phases of eDNA sequencing-based community science projects.
We propose three avenues for future work with ranacapa. First, we plan to use ranacapa as the primary tool to present eDNA results from hundreds of samples sequenced by the CALeDNA community science program. Second, ranacapa is being integrated into the upcoming undergraduate curriculum module “Pipeline for Undergraduate Microbiome Analysis”, which will be an open-source, comprehensive suite of analysis and data visualization tools for undergraduate researchers. Finally, in the long-term, we believe there is great promise in linking ranacapa with packages that connect with APIs of online biodiversity databases (e.g. Taxize (Chamberlain & Szöcs, 2013), rinat (Barve & Hart, 2017)). This will help users explore a much wider range of biodiversity questions, for example, by programmatically asking whether their samples include invasive species that are absent from other nearby sites. In sum, tools like ranacapa that allow non-technical audiences to easily interact with results from eDNA sequencing studies have great potential to engage community partners with a wide range of backgrounds and interests in primary research.
• A Shiny app, including a dataset generated for demonstrations, is available at https://gauravsk.shinyapps.io/ranacapa
• Source code is available from GitHub: https://github.com/gauravsk/ranacapa
• Archived source code at time of publication: http://dx.doi.org/10.5281/zenodo.1464285 (Kandlikar & Cowen, 2018)
• Software license (GPL-3)
Datasets used for the Use cases are available from Figshare:
Dataset 1: Taxon table and metadata file for Channel Islands eDNA samples (mitochondrial 12S and CO1 metabarcodes sequenced) https://doi.org/10.6084/m9.figshare.7199477.v1 (Kandlikar et al., 2018a)
Dataset 2: Taxon table and metadata file for Santa Monica Mountains eDNA samples (16S and plant-ITS metabarcodes sequenced) https://doi.org/10.6084/m9.figshare.7199510.v1 (Kandlikar et al., 2018b)
Both datasets are available under a CC-BY 4.0 license
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)