Keywords
Protein 53, SNPs, in silico Analysis, Esophageal cancer, Sudan.
Protein 53, SNPs, in silico Analysis, Esophageal cancer, Sudan.
Esophageal cancer is considered one of the eight most common cancers throughout the world, and is also one of the most fatal cancers, taking into account its aggressiveness and reduced survival rate. Because of its poor prognosis with 5-year survival rates ranging between 10–13%, it ranks sixth among all cancers in mortality rate1–4.
Knockout of TP53 in mice leads to the development of different tumors, including lymphomas, sarcomas adenocarcinoma and benign tumors such as hemangioma, before they reach 6 month of age5.
TP53 gene encodes a tumor suppressor protein which plays an important role inside the cell especially in DNA transcription and repair, senescence, apoptosis, tumor suppression, treatment response and also the response to changes in metabolism6,7. Protein domains represent independently folding units of protein with a size between 40 to 200 amino acids. Human p53 protein contains three domains; transcriptional activation, DNA binding, and oligomerization domains. These domains are edged by a connecting region. A proline-rich region links the transcriptional activation and DNA binding domains, a second proline-rich region links the DNA binding and oligomerization domains and a basic region form the C-terminus of the protein8. The evolutionarily highly conserved core domain (amino acids ~100 to ~300) is involved in sequence-specific binding to promoters of p53-regulated genes9.
Single nucleotide polymorphisms (SNP) are a significant type of genetic variation commonly detected in the human genome. SNPs occur in non-coding regions as well as in coding regions of the genome10,11. A total of 336,845,724 SNPs have been identified in humans so far, and have been deposited in NCBI dbSNP. The human TP53 gene has 3115 identified SNPs. SNPs arise in coding regions may cause an amino acid change in the corresponding protein and in such case it is called as non-synonymous SNP (nsSNP) or may not change the amino acid and here it is called a synonymous SNP (sSNP); these nsSNPs change the protein structure and hence its function, causing a specific disease12,13.
Recently a number of articles have demonstrated the association of SNPs in the TP53 gene with different cancer types, but in silico analysis has not yet been discussed on the functional, interactional and structural aspects of different types of SNPs in this gene. In the current study, we used different bioinformatics prediction tools and databases for analysis of these SNPs in TP53 gene. As a significant number of mutations have an impact on protein stability and interactions with the corresponding proteins, we also offered a structural model of the mutant protein. Here in this study the main objective is to detect mutations of TP53 gene focusing on exons 5 to 8 among esophageal cancer patients as these has been reported as the most mutated exons in this gene14.
Sections of 30–40 µm thickness from 50 formalin fixed paraffin embedded (FFPE) tissue samples were obtained from esophageal cancer patients representing different hospitals and clinics in Khartoum State, Sudan, from July 2013 to June 2017. All patients have been previously diagnosed with squamous cell carcinoma (SCC) and adenocarcinoma (AC).
Genomic DNA for PCR analysis was extracted from FFPE tissue blocks. Using commercial DNA extraction kits for fast isolation of genomic DNA from FFPE samples as per manufacturer’s instructions. Extraction procedure is based on combination of an efficient lysis step with a subsequent binding of genomic DNA on a Spin Filter surface followed by washing of the bound DNA and finally eluting of the DNA (845-BP-0020250, black PREP FFPE DNA Kit, Analytik Jena Company).
For amplification of exon 5, 6,7 and 8 of TP53 gene, four pairs of primers (catalogue numbers: 171002-009_D5, 171002-009_D6, 171002-009_D7, 171002-009_D8, 171002-009_D9, 171002-009_D10, 171002-009_D11, 171002-009_D12; Macrogen, Korea) were used15,16 (Table 1). A total of 2–5 µl of genomic DNA, 0.5 µl forward and 0.5 µl reverse primer and 25 μl double distilled water (DDW) was combined to make up the final reaction volume. The mixture was amplified using Heal force thermal cycler (Model No: K960) with the following amplification conditions; 95°C for 5 min, followed by 37 cycles at 95°C for 45 sec, primer-specific annealing temperature for 45 sec, 72°C for 45 sec and a final extension at 72°C for 5 min. 5 μl of the PCR products were applied on 2 % agarose gel and remaining PCR products were sequenced by BGI company (China).
The SNP information SNP ID, mRNA accession number NM_000546, and Protein accession number NP_000537 of the human TP53 gene used in our computational analysis were retrieved from the National Center for Biotechnology Information (NCBI) database and catalogue of somatic mutation in cancer (COSMIC) database (TP53_ENST00000269305). The nucleotide and amino acid sequence of the p53 protein were obtained and investigated using nucleotide (NG_017013), Gene (Gene ID: 7157) database NCBI and UniProt database (P04637).
Codon code aligner. Sequences were assembled into contigs end clipped and edited using Codon Code Aligner software version 8.0.1 (Dedham, MA, USA). Sequence data are available at GenBank under accession numbers MH366303 to MH36648317.
SIFT Program. SIFT (Sorting Intolerant from Tolerant) tool uses sequence homology to calculate the probability of affecting protein function in case of amino acid change. It uses the concept of evolutionarily conserved regions which is less tolerant to mutations, and therefore amino acid change or frame shift mutations in these regions are expected to affect protein function the most. SIFT tool works by introducing a query protein into SIFT program to be searched against protein database aligned with homologous protein sequences. Then the program calculates SIFT score based on amino acid changes in that position. A SIFT score ranges from 0 to 1. Score less than 0.05 is predicted to affect protein function and considered functionally deleterious, whereas any score more than or equal to 0.05 represents a neutral substitution18–20.
PolyPhen -2. PolyPhen-2 (Polymorphism Phenotyping version 2) is a structural and functional predicting tool that predicts the effect of an amino acid change on protein characteristics based on SNPs functional annotations, protein structural properties with sequence annotation, and finally predict if the coding non-synonymous SNPs are considered damaging or not21,22.
PolyPhen-2 workflow requires protein sequence, mutational position, and substitution. The PolyPhen output is represented with a score that ranges from 0 to 1, with zero score indicating a neutral effect of amino acid substitution on protein function and a higher score representing a mutation that is more likely to be damaging23.
I-Mutant 3.0. I-Mutant 3.0 is a support vector machine (SVM) based tool, which was used to calculate the stability changes of specific SNP upon protein sequence. Information of wild and mutated residue, protein sequence, temperature, and pH was used as input parameters to this server, and finally, the outputs reports if a point mutation is stable or not. The program categorizes the prediction into: neutral mutation (DDG = 0.5 kcal/mol), large decrease of stability (0.5 kcal/mol). The output is a free Gibbs energy change value (ΔΔG) of protein before and after mutation24–27.
PhD-SNP. PhD-SNP (Predictor of Human Deleterious Single Nucleotide Polymorphisms) software is a prediction tool that predicts disease association of nsSNP by dividing those SNPs into disease-related or neutral polymorphism based on a score ranged from (0-1); SNPs with a score above 0.5 are considered disease associated according to the program algorithm. PhD-SNP outputs depend on a number of sequences aligned, conservation index of SNP position, frequencies of wild and mutant residues19,20,28.
Project HOPE. Structural and biochemical analysis for mutations was accomplished using Project HOPE is a web-server used to give a comprehensive report on the effect of the specific mutation on the 3D structure of the native protein and the variant model using different software and sources. The user can submit a protein sequence or an accession number of specific protein after specifying the wild-type residue and the new mutant form to create the report29,30.
Mutation Taster. Mutation Taster calculates the pathogenic consequences of variations in DNA sequence. It predicts the functional impact of amino acid alterations, intronic and synonymous substitutions, in addition to INDEL mutations and variants covering intron-exon connection region. Mutation Taster prediction system divides alterations as; Disease-causing: which is probably deleterious, Disease-causing automatic: the alteration here is known to be deleterious, Polymorphism: probably harmless alteration and polymorphism automatic: known to be harmless31–33.
FATHMM. FATHMM (Functional Analysis Through Hidden Markov Models) is a web-server predicts the functional significances of both coding and non-coding variants. We selected the cancer option to display predictions that can distinguish between cancer-promoting/driver mutations and other germline polymorphisms. It uses a default prediction threshold of -0.75. Predictions with scores less than this indicate that the mutation is potentially cancer associated34.
Esophageal squamous cell carcinoma cases represent 43 (86%) of all cases, whereas adenocarcinoma made up 7 cases (14%). Mutation analysis results demonstrate a higher TP53 mutation rate in esophageal adenocarcinoma compared to squamous cell carcinoma. This were illustrated in (Table 2) in the results section which describe Histopathological diagnosis, mutational status and exons affected in esophageal cancer patients.
Distribution of TP53 coding synonymous SNPs (sSNPs), coding non-synonymous SNPs (nsSNPs) and INDEL through exon 5-8 in P53 gene were represented by (Figure 1).
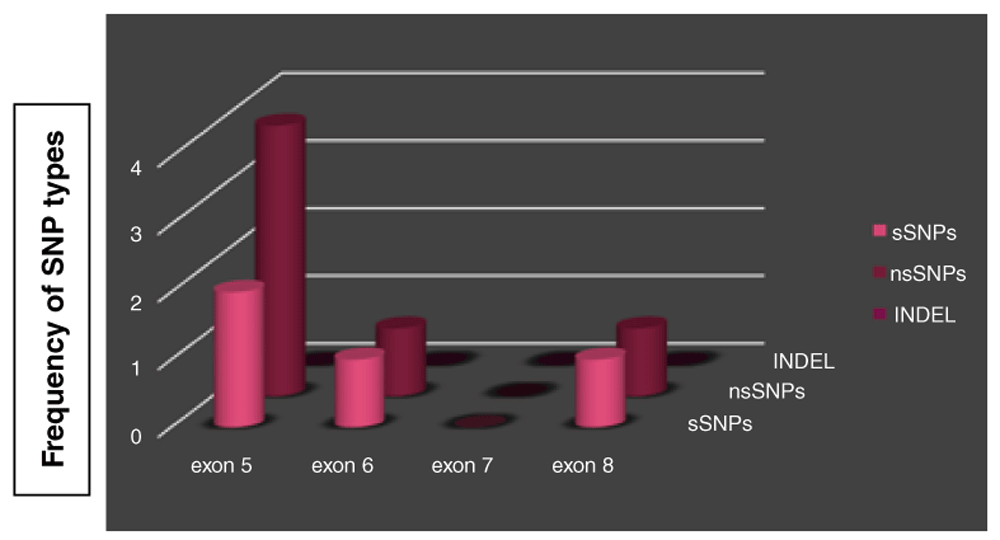
TP53 gene SNPs were found in 20 out of 50 sample of esophageal carcinomas (40%). Six out of ten SNPs (60%) were missense SNPs leading to amino acid substitution, four SNPs (40%) were silent mutations without any amino acid change. Six out of ten (60%) TP53 SNPs occurred in exon 5 at the following codons 160, 161 (twice), 163, 164, 175. Three of them were missense and three were silent. Two SNPs (20%) were located in exon 6 at codon 215 (missense) and 222 (silent). Two SNPs (20%) were present in exon 8 at codon 298 (missense) and 305 (silent).
15 out of 43 (34.9%) SSC samples were found to be mutated, 13/15 (86.7%) of them existed in exon 5, 2/15 (13.3 %) in exon 6 and 1/15 (6.7%) in exon 8. Whereas adenocarcinoma had a higher rate of mutations 4/7 (57.1%) with 100% of SNPs occurring in exon 5.
The percentage of deleterious nsSNPs predicted by SIFT and PolyPhen was 66.7% (Figure 2), those SNPs were A161D, K164E, R175P and S215N according to SIFT and PolyPhen (Table 3–Table 4). I-Mutant suite also give the same percentage for deleterious nsSNPs which is 66.7% (Table 5) but in case of using I-Mutant suite the predicted SNPs to be deleterious were, A161D, R175P, S215N and M160V (Table 6). The PhD-SNP report defines 5/6 (83.3%) of nsSNPs as disease related polymorphism (Table 7).
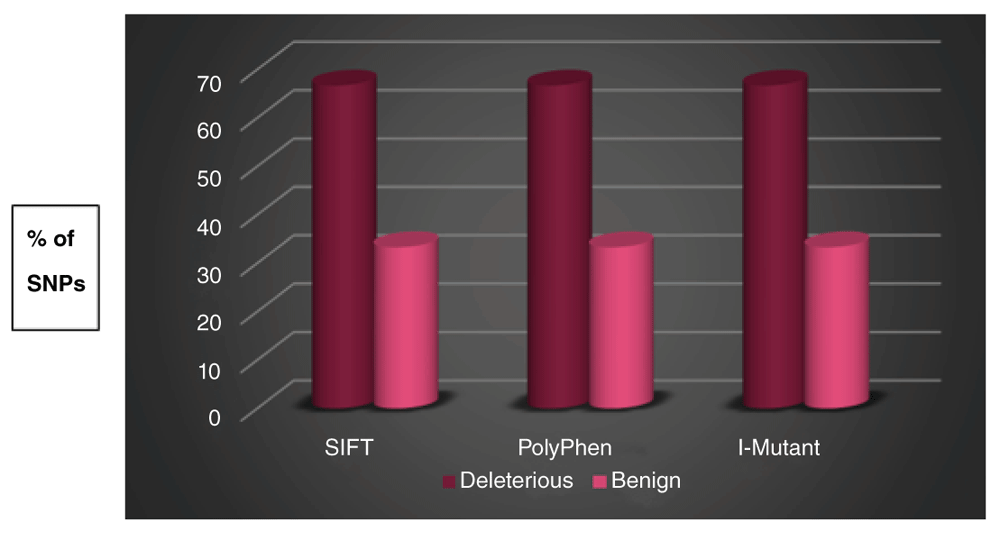
The magenta cylindrical bar indicates the percentage of nsSNPs that were found to be deleterious by SIFT, damaging (Possibly/Probably) by PolyPhen, and largely unstable by I-Mutant Suite. The pink cylinder indicates the percentage of nsSNPs that were found to be tolerated by SIFT, benign by PolyPhen, and largely stable/neutral by I-Mutant Suite.
| Prediction Result | SIFT | PolyPhen | I Mutant 3.0 | |||
|---|---|---|---|---|---|---|
| No. of nsSNPs | % | No. of nsSNPs | % | No. of nsSNPs | % | |
| Deleterious | 4 | 66.7 % | 4 | 66.7 % | 4 | 66.7 % |
| Tolerated | 2 | 33.3 % | 2 | 33.3 % | 2 | 33.3 % |
| Total | 6 | 100% | 6 | 100% | 6 | 100% |
The results reveal that SNPs in positions E298Q were predicted to be a neutral polymorphism which represent 10% of all mutation detected. All other SNPs M160V, A161D, A161A, Y163Y, K164E, R175P, S215N, P222P, and K305K representing 90% of all SNPs were predicted to be disease related according to MutationTaster software (Table 8).
| Localization | Patient affected | alleles | AA Change | Position | SNP ID | Type | Significance* |
|---|---|---|---|---|---|---|---|
| Exon 5 | 14 | A/G | K164E | 7675122 | rs879254249 | Missense | Disease causing |
| Exon 5 | 1 | C/A | A161D | 7675130 | rs1064795691 | Missense | Disease causing |
| Exon 5 | 9 | A/G | M160V | 7675134 | rs377274728 | Missense | Disease causing |
| Exon 5 | 2 | C/T | Y163Y | 7675123 | COSM44391 | Coding-synonymous | Disease causing |
| Exon 5 | 1 | G/C | R175P | 7675088 | COSM45416 | Missense | Disease causing |
| Exon 5 | 1 | C/T | A161A | 7675129 | COSM44119 | Coding-synonymous | Disease causing |
| Exon 6 | 1 | G/A | S215N | 7674887 | rs587782177 | Missense | Disease causing |
| Exon 6 | 1 | G/C | P222P | 7674865 | COSM43924 | Coding-synonymous | Disease causing |
| Exon 8 | 1 | A/T | E298Q | 7673728 | Novel | Missense | Polymorphism |
| Exon 8 | 1 | G/A | K305K | 7673705 | COSM46382 | Coding-synonymous | Disease causing |
Frequency of SNPs among different samples was shown in details in (Table 9). FATHMM server; cancer association predictions result of the non-synonymous changes found in TP53 gene exon 5-8 were demonstrated in (Table 10).
| COSMIC ID | Amino acid change | Prediction | Score |
|---|---|---|---|
| COSM44328 | M160V | CANCER | -9.03 |
| COSM44391 | A161D | CANCER | -9.34 |
| COSM10762 | K164E | CANCER | -9.12 |
| COSM45416 | R175P | CANCER | -9.93 |
| COSM44093 | S215N | CANCER | -9.58 |
| COSM45938 | E298Q | CANCER | -8.11 |
Different mutations with their position, wild type and mutant form in addition to alignment and chromatogram were illustrated in (Figure 3–Figure 8).
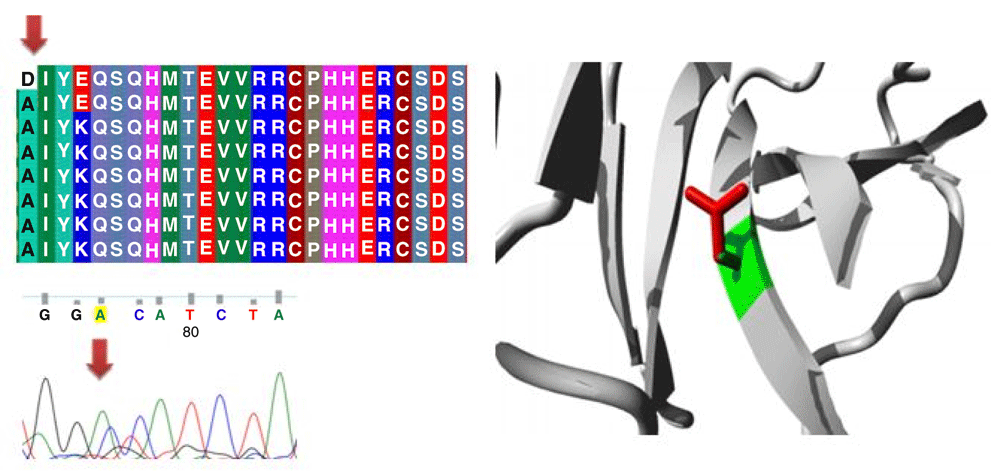
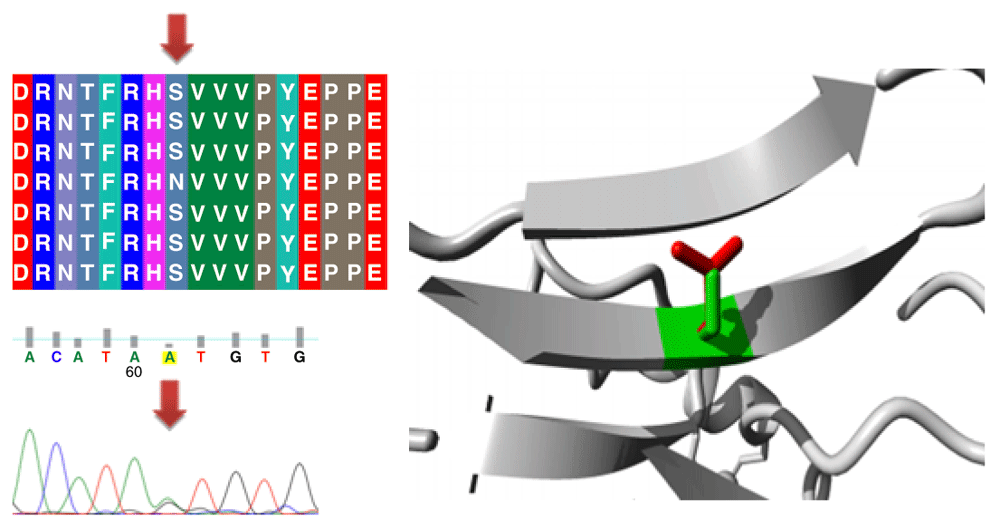
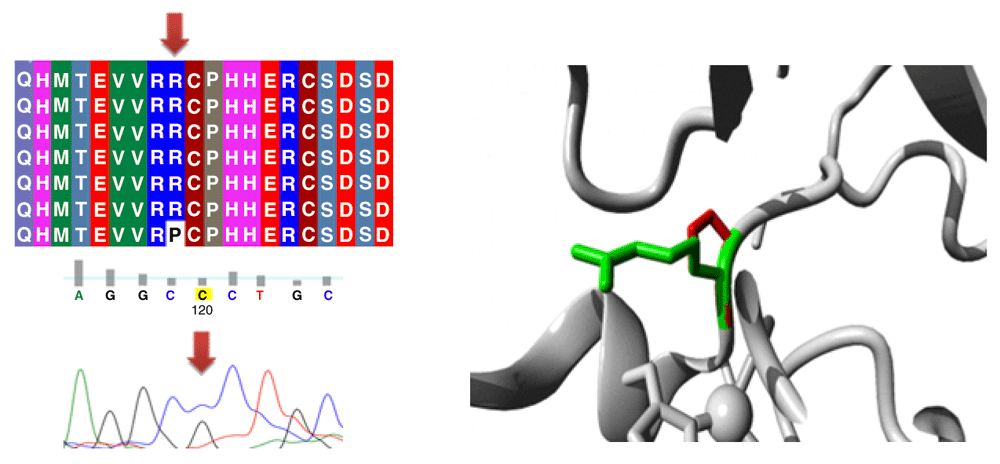
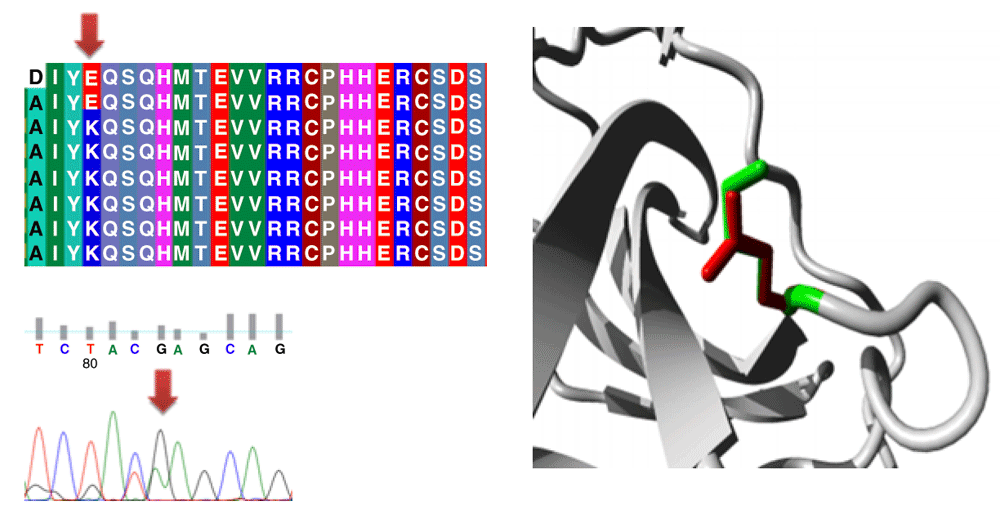

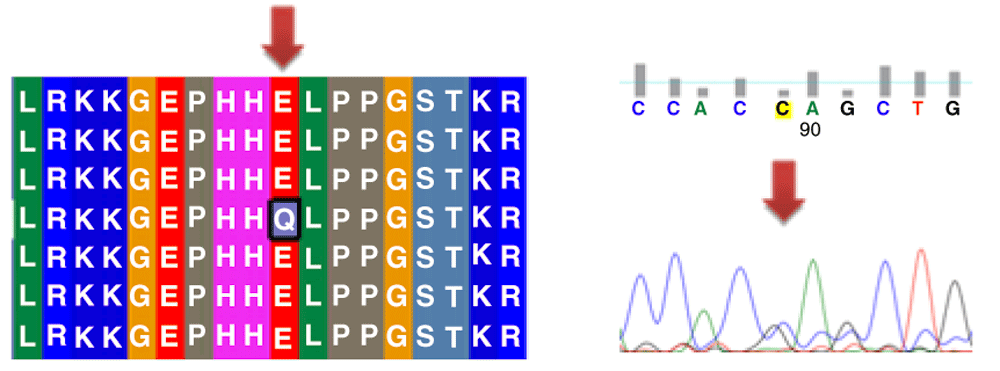
Mutations of TP53 gene can lead to loss of functional characteristics in tumor cells, as mutant TP53 may not play the assigned role in repairing cellular machinery leading to a loss of normal function and, subsequently, cells with mutant gene may express uncontrolled replication which leads to accumulation of protein 5313.
There were 8 (40%) males with TP53 gene mutations and 12 (60%) females. TP53 mutations were found in 22.9% of esophageal SCCs and 14.3% of esophageal ACs in a study done by Zheng et al.35 here the ratio was (34.9% vs. 57.1%) respectively. Sample size may be the main factor affecting the result.
Shi et al.36 in Henan province, China showed that the p53 mutations were detected in 30 out of 43 (70%) SCC cases but in this study it represents 16 out of 43 (37.2%); this difference can be attributed to different factors e.g. geographical zone differences or type of exons studied as China has a high-incidence of esophageal carcinoma. Another study in Henan province, China conducted by Li-Ya15 and his colleagues stated that p53 mutations were detected in 40.9% of their esophageal cancer specimens and this is compatible with our finding on mutations being detected in 20 out of 50 specimens of esophageal carcinomas which represent 40%.
In a study conducted by Zheng et al.35 Qiqihar City, China, a higher mutation rate was found in SCC samples compared to AC samples (31.4% vs. 21.4%, respectively). And this partially differs from our result which found 4 out of 7 (57.1%) of adenocarcinoma patients had mutations which revealed higher mutation rate among AC samples, and this result may differ if had a larger sample size with more adenocarcinoma cases.
Exon 5 of the TP53 gene was observed to be the most mutated exon among the other exons investigated here in this study, with 17/20 (85%) of all mutations detected found in exon 5, 2/20 (10%) in exon 6 and 1/20 (5%) in exon 8, while exon 7 showed no mutations. Uchino et al.37 results for mutation distribution across exons was 39.3% in exon 5, 32.1% in exon 6, 17.9% in exon 7 and (10.7%) forexon 8 mutations. Which has partly agreed with our result in exon 5 having the higher mutation rate in addition to reasonable differences in other exons rate of mutation, and this can be attributed to the small sample size used in this study.
A total of 10% of all detected mutations were classified as neutral polymorphism, while 90% were considered disease-causing according to Mutation Taster, considered high rate of disease-causing mutations. This rate is lesser using other software due to different algorithms used by any one of them, their linked databases, and characteristics of the different software.
Mutation of exon 5 in p53 gene were the most frequent in esophageal cancer. Genomic results have identified a high TP53 mutation rate in esophageal Adenocarcinoma compared to squamous cell carcinoma.
This study was approved by the Institutional Ethics Committee, Sudan University of Science and Technology (reference number for the ethical committee is DSR-IEC-13-05). Patients consent cannot be obtained because most of the patients were dead and the rest cannot be traced due to lack of contact data. Therefore all samples and medical data used in this study have been irreversibly anonymized to ensure patients privacy.
TP53 sequences for this study has been submitted to Banklt NCBI and had been assigned the accession numbers MH366303 to MH366483. The sequences are available as 4 PopSet entries for exons 5–8:
Exon 5: 1472901613
Exon 6: 1472901713
Exon 7: 1472901809
Exon 8: 1472901897
TP53 sequence results were submitted in a zipped file. These sequencing results as received from BGI Company (China) include 50 esophageal cancer patients in this study using the four sets of primers for exon 5, 6, 7 and 8. Sequencing files that needs to be viewed using FinchTV and Notepad file.
Dataset 1: Patients information, clinical data, and histological findings of all patients were available in Excel format. 10.5256/f1000research.15534.d21973938
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)