Keywords
Carotid body, Heme-oxygenase, Gasotransmitter, NADH dehydrogenase Fe-S protein 2
Carotid body, Heme-oxygenase, Gasotransmitter, NADH dehydrogenase Fe-S protein 2
Systemic hypoxia, which arises from decreased oxygen (O2) levels in the arterial blood, is a fundamental physiological stimulus. The duration of hypoxia can be acute, ranging from seconds to minutes, or chronic, lasting hours to days. Acute hypoxia evokes rapid changes in the cardiorespiratory systems to ensure optimal O2 delivery to tissues. Cardiorespiratory responses to acute hypoxia are primarily reflexive in nature, initiated by sensory organs located in the carotid artery and aorta. Carotid bodies (CBs), which reside at the bifurcation of the common carotid arteries, are the major sensory organs for monitoring arterial blood O2 levels1. Although structures similar to CBs are seen at the aortic arch and in the abdominal arteries, much of the information on the mechanisms of hypoxic sensing has come from studies on the CB1. Here, we present studies reported in the past five years on the roles for heme oxygenase-2 (HO-2), a carbon monoxide (CO)-synthesizing enzyme, and NDUFS2, a mitochondrial complex I subunit, in hypoxic sensing by the CB.
The CB receives sensory innervation from the carotid sinus nerve, whose cell bodies reside in the petrosal ganglion. Under basal conditions (arterial blood pO2 of about 100 mmHg), sensory nerve discharge (that is, frequency of action potentials) is low. In response to even a modest decrease in arterial blood pO2 from 100 to 80 mmHg, the sensory discharge increases and the response is fast, occurring within a few seconds after the onset of hypoxia1. The increased sensory discharge is non-adapting and is maintained during the entire duration of hypoxia2 or may progressively increase during sustained hypoxia, lasting several hours3. The exquisite sensitivity and the speed of the response with little or no adaptation are the unique features of hypoxic sensing by the CB. The increased CB sensory nerve activity is relayed to brainstem neurons, leading to reflex stimulation of breathing and blood pressure (CB chemo reflex)1.
The CB tissue is made of two major cell types: type I cells (also called glomus cells), which are of neuronal origin, and type II cells, which resemble glial cells of the nervous system. Type I cells along with the nearby sensory nerve ending function as a “sensory unit”1. Stimulus response of breathing to graded hypoxia parallels the CB sensory nerve activity1. Consequently, carotid sinus nerve activity is measured as an index of CB hypoxic sensing1. Type I cell responses to acute hypoxia are measured by monitoring exocytosis and changes in [Ca2+]i and K+ channel conductance1. Although type I cells respond to hypoxia with elevated [Ca2+]i or K+ channel inhibition (or both), they are not always reflected in the sensory nerve activity4,5, which is essential for evoking the physiologically important CB chemo reflex. Therefore, it is necessary to corroborate the cellular responses to hypoxia with the sensory nerve discharge for assessing the physiological relevance of CB hypoxic sensing.
The consensus is that hypoxia inhibits certain K+ channels in type I cells and the resulting depolarization leads to Ca2+-dependent release of neurotransmitter or neurotransmitters, which stimulate the nearby sensory nerve ending, leading to increased sensory discharge1. The roles for K+ channels and AMP kinase (AMPK) in CB hypoxic sensing have been discussed in detail elsewhere1,6,7 and will not be presented in this commentary. The following section presents studies conducted in the past five years that have provided novel insights into the roles for HO-2 and mitochondrial complex subunit NDUFS2 in hypoxic sensing by the CB.
Type I cells express HO-2-like immunoreactivity8. HO-2 is remarkably sensitive to O2 availability, and graded hypoxia progressively decreases CO production in the CB9. Reduced CO production by hypoxia is also seen in HEK-293 cells with heterologous expression of HO-29, suggesting that HO-2 is inherently sensitive to O2. HO-2 binds to O2 with low affinity with an apparent Km of 65 ± 5 mmHg (about 80 µM). The O2 sensitivity of HO-2 is due to Cys265 and Cys282 residues in the heme regulatory motif9. Intact Cys265 and Cys282 residues lower the affinity of HO-2 for O2 and thereby enable the enzyme to transduce changes in O2 into changes in CO production. Substituting Cys265 and Cys282 with alanine allows the HO-2 to bind to O2 with high affinity9.
It was proposed, based on the findings that CO inhibits CB sensory nerve excitation by hypoxia8–10 and that hypoxia reduces CO production in a stimulus-dependent way9, that low sensory nerve activity during normoxia is due to high CO levels inhibiting CB sensory nerve activity but that hypoxia, by reducing CO production, relieves the inhibition and thereby increases the sensory nerve discharge8. Recent studies determined how CO inhibits CB sensory nerve activity under normoxia9,10. Type I cells also express cystathionine gamma-lyase (CSE), an enzyme catalyzing hydrogen sulfide (H2S) production11. H2S is a potent stimulator of CB sensory nerve activity in rats, mice, rabbits, and cats11–13. During normoxia, H2S levels are low and hypoxia increases H2S levels in a stimulus-dependent manner11. The increased H2S production is not due to inherent O2 sensitivity of CSE; rather, it is due to changes in CO production9. High CO levels during normoxia inhibit CSE-derived H2S generation through protein kinase G (PKG)-dependent phosphorylation of Ser377 of CSE, and reduced CO generation during hypoxia relieves the inhibition of CSE, leading to increased H2S generation in the CB9. CSE inhibitors and CSE knockout mice exhibit impaired type I cell and sensory nerve and breathing response to hypoxia11,14. These findings suggest that low sensory discharge during normoxia is due to inhibition of H2S generation by high levels of HO-2-derived CO but that the increased sensory nerve activity by hypoxia is due to relieving inhibition of H2S synthesis by CO (Figure 1).
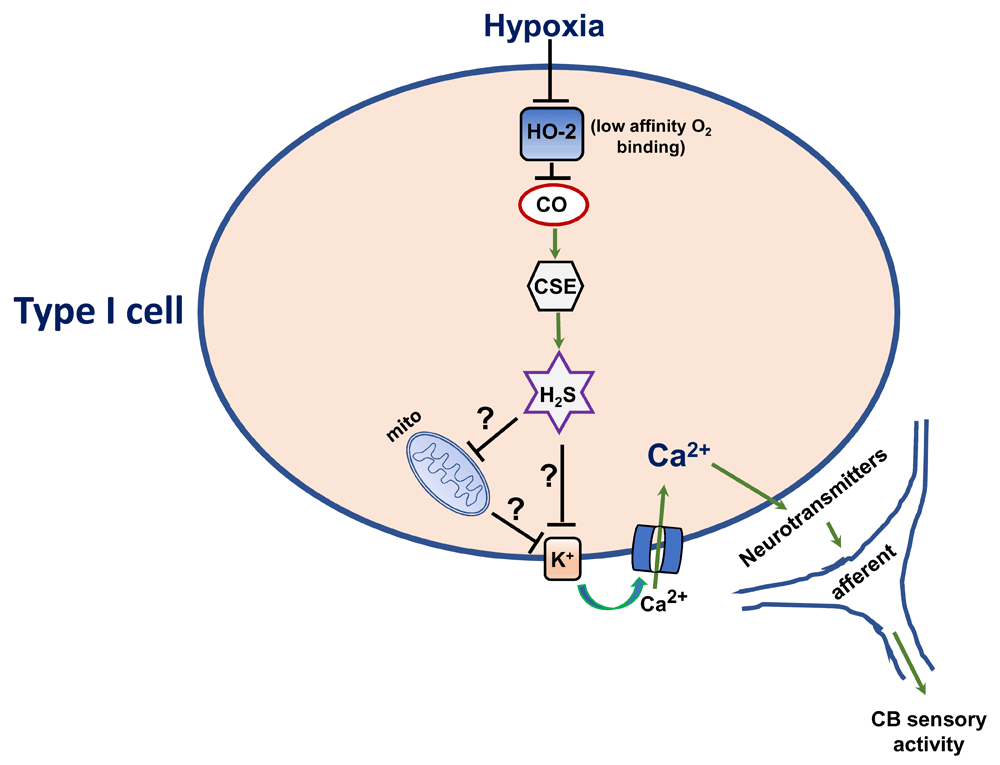
Ca2+, calcium channel; CO, carbon monoxide; CSE, cystathionine gamma-lyase; H2S, hydrogen sulfide; K+, potassium channel; mito, mitochondria.
Genetic disruption of HO-2 increases baseline CB sensory nerve activity and elevates H2S levels under normoxia, a phenotype similar to hypoxia9. However, hypoxia further increased CB sensory activity and elevated H2S levels in HO-2 null CBs, indicating the existence of a redundant hypoxic sensing mechanism (or mechanisms). This redundant hypoxic sensing was due to compensatory upregulation of neuronal nitric oxide synthase (nNOS) in type I cells of HO-2 null mice9. nNOS, like HO-2, also binds to O2 with low affinity, and nitric oxide (a product of nNOS), like CO, also inhibits CSE-derived H2S through PKG signaling9. Blockade of nNOS in HO-2 null mice renders CBs completely insensitive to hypoxia9. These findings suggest that, in the absence of HO-2, nNOS contributes to CB hypoxic sensing by regulating CSE-derived H2S production9.
A recent study examined the role for HO-2–CO signaling in CB hypoxic sensing of three genetically distinct rat strains that are commonly used in experimental research10. As compared with Sprague-Dawley rat CB, Brown-Norway (BN) rat CB showed markedly attenuated sensory nerve and type I cell responses to hypoxia, and this phenotype was associated with higher CO and lower H2S levels in the glomus tissue. The elevated CO levels in the BN rat CB were due to high affinity of HO-2 to its substrate hemin10. The attenuated CB response to hypoxia is associated with a blunted chemo reflex. The CB chemo reflex is essential for ventilatory adaptations to high-altitude hypoxia15,16. BN rats exposed to hypobaric hypoxia showed severe pulmonary edema10, which is a sign of chronic mountain sickness. Treating BN rats with a HO inhibitor restored the hypoxic response of the CB and prevented pulmonary edema caused by hypobaric hypoxia10.
In contrast to BN rat CBs, CBs of spontaneous hypertensive (SH) rats showed heightened sensitivity to hypoxia, and this phenotype is associated with low CO levels and high H2S levels in the CB10. The low CO levels in SH rat CBs were due to low hemin affinity of HO-210. Current evidence suggests that a hyperactive CB chemo reflex is a major contributor of hypertension in SH rats17,18. Systemic administration of a CSE inhibitor normalized CB sensory nerve and type I cell responses to hypoxia and reduced hypertension10. Although chronic ablation of CB also reduced hypertension to the same level as seen with a CSE inhibitor, combined CB ablation and CSE inhibitor treatment had no further effect on blood pressure10. These findings suggest that CO-regulated H2S contributes to a hyperactive CB in SH rats. It has long been known that the CB chemo reflex exhibits substantial inter-individual variations in humans and experimental animals19–21. Studies on BN and SH rats indicate that inter-individual variations in chemo reflex are due in part to variations in HO-2–CO signaling in the CB.
A recent study suggests that HO-2–CO signaling in the CB also plays an important role in the pathology of sleep apnea (SA), which is a highly prevalent respiratory disease22. SA is characterized by episodic cessation of breathing leading to chronic intermittent hypoxia (CIH). Patients with SA and CIH-exposed rodents exhibit a heightened CB chemo reflex, leading to chronic elevation of sympathetic nerve activity and hypertension23–26. Rodents exposed to CIH exhibit elevated reactive oxygen species (ROS) levels in the CB27,28. Peng et al. showed that ROS inhibit CO generation by HO-2 by acting on the Cys265 residue in the heme regulatory motif, thereby increasing H2S levels in the CB22. Pharmacological or genetic blockade of CSE-derived H2S prevents CIH-induced CB hyperactivity, sympathetic nerve excitation, and hypertension22. Collectively, these studies suggest that disrupted HO-2–CO signaling in the CB leads to dire physiological consequences.
Although the studies described above suggest that CO-regulated H2S is an important mediator of CB hypoxic sensing, a recent study questioned this possibility. Kim et al.29 reported that inhibitors of H2S synthesis had no effect on [Ca2+]i and TASK K+ channel responses of type I cells to anoxia (pO2 of about 5 mmHg). Peng et al.5 re-examined the role for CSE-derived H2S in the CB sensory nerve and type I cell [Ca2+]i responses to hypoxia (pO2 of about 37 mmHg) and anoxia (pO2 of about 5 mmHg). The authors found that hypoxia increased H2S levels in the CB, stimulated sensory nerve activity, and elevated [Ca2+]i in type I cells and all of these responses were blocked by a CSE inhibitor and in CSE knockout mice. In striking contrast, anoxia, though producing very low pO2, had no effect on H2S levels in the CB and produced only a weak CB sensory nerve excitation as compared with hypoxia. CB sensory and type I cell responses to anoxia were unaffected by CSE inhibitors and in CSE knockout mice. Moreover, anoxia (100% N2) depressed breathing whereas hypoxia (12% O2) stimulated breathing5. CSE knockout mice showed an absence of breathing stimulation by hypoxia, whereas the depressed breathing by anoxia was unaffected in these mice5. These findings suggest that hypoxia and anoxia are not the same stimuli for studying CB physiology and that HO-2–CO-regulated H2S mediates CB response to “physiologically relevant” hypoxia but not anoxia.
Mitochondrial electron transport chain (ETC) inhibitors mimic the effects of hypoxia on CB sensory nerve activity1 and type I cells30–33. Mills and Jöbsis34 reported that CBs express a putative cytochrome aa3 with two O2 affinities: one with high and another with low affinity for O2. Based on spectral analysis, subsequent studies suggested that CBs express a cytochrome, which is half-reduced at a pO2 of 60 to 80 mmHg, and this cytochrome is not expressed in either the superior cervical or the nodose ganglion35,36. Acute hypoxia increased the NADH/NAD ratio and decreased mitochondrial membrane potential in type I cells, and these effects were not seen in other non-chemoreceptor tissues such as dorsal root ganglion37. These studies led to the suggestion that mitochondrial ETC participates in CB hypoxic sensing.
Rotenone, an inhibitor of the mitochondrial complex I, selectively blocks the type I cell response to hypoxia32. NDUFS2 is a component of the complex I, which binds to ubiquinone38–40. Recent studies examined the role for complex I in CB hypoxic sensing in mice with targeted deletion of Ndufs2 in tyrosine hydroxylase-positive (TH+) cells such as type I cells41. Mice lacking NDUFS2 in TH+ cells showed an absence of breathing stimulation by hypoxia and of hypoxia-evoked exocytosis and K+ channel inhibition in type I cells. However, type I cell responses to severe hypercapnia (20% CO2) were intact41. Given that these mice have a deletion of NDUFS2 since birth, the lack of breathing response to hypoxia might be secondary to metabolic adaptations during development. Additional studies were performed on adult (2-month-old) mice with conditional knockout of Ndufs2 (ESR-NDUFS2 mice). Like the TH-NDUFS2 mice, mice with conditional knockdown of Ndufs2 showed an absence of stimulation of breathing as well as type I cell responses to hypoxia41. The lack of cellular responses to hypoxia was associated with decreased complex I activity, complex I formation, and complex I-dependent O2 consumption, whereas the functions of other mitochondrial complexes were intact. These studies suggest that NDUFS2 of the mitochondrial complex I contributes to hypoxic sensing by the CB.
How might NDUFS2 confer hypoxic sensitivity on type I cells? Mitochondrial ETC-generated ROS have been implicated in pulmonary artery myocyte responses to acute hypoxia42–44. Acute hypoxia increased ROS in wild-type type I cell cytosol and mitochondrial intermembrane space, and these responses were attenuated in NDUFS2 null type I cells41. Intracellular application of H2O2, like hypoxia, inhibited background K+ currents in type I cells41. These findings led to the suggestion that inhibition of NDUFS2 leads to an increase in ROS production, which in turn (by inhibiting K+ currents) leads to depolarization of type I cells by hypoxia (Figure 2).
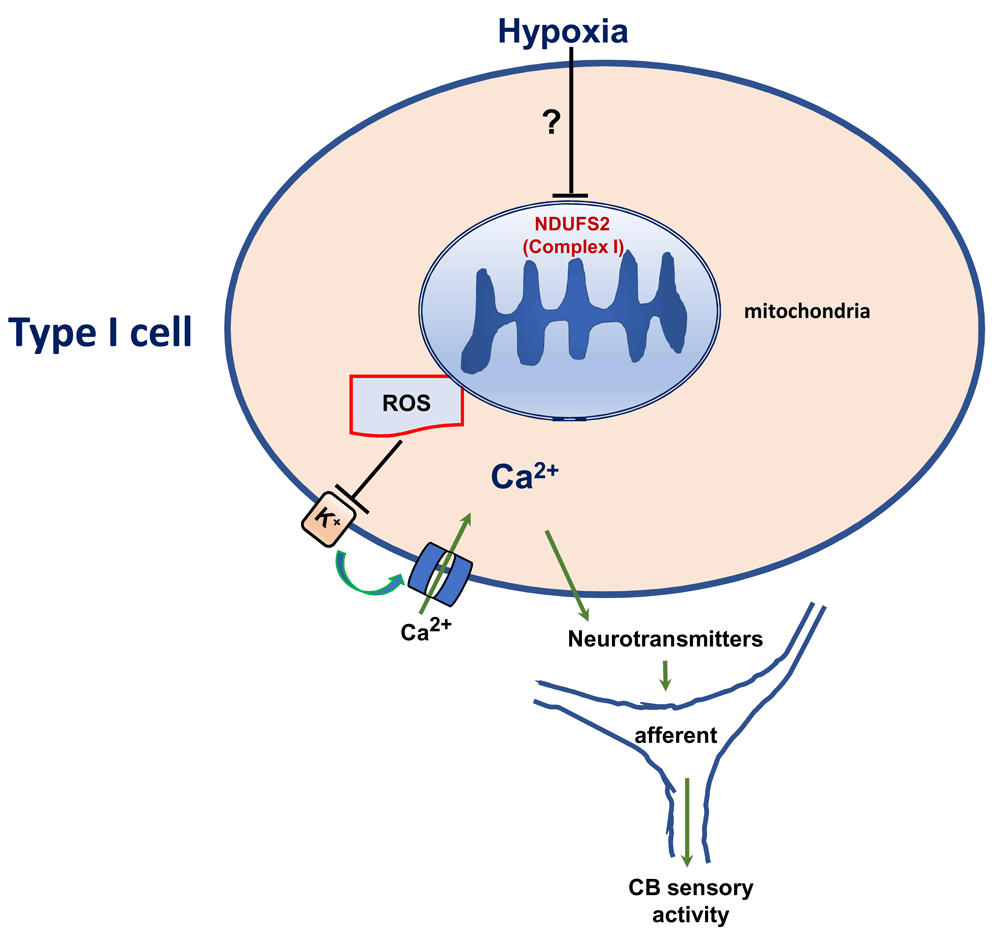
Ca2+, calcium channel; K+, potassium channel; ROS, reactive oxygen species.
It has long been thought that an O2 sensor or sensors in type I cells initiate hypoxic sensing in the CB1,45,46. To be considered an O2 sensor, a molecule should satisfy certain criteria, namely (a) its presence in type I cells, (b) low-affinity binding to O2, (c) altered function by hypoxia should initiate signaling events leading to increased CB sensory nerve activity, and (d) loss of CB hypoxic sensing by disrupting its function. HO-2 satisfies the proposed criteria for an O2 sensor in the CB and contributes to CB sensory excitation by regulating H2S production through O2-dependent CO production. However, further studies are needed to delineate the cellular mechanism(s) underlying CB activation by H2S. H2S donors, like hypoxia, depolarize47 and inhibit K+ channel conductance in type I cells12,48 and increase NADH auto-fluorescence in type I cells, an effect attributed to the inhibition of mitochondrial ETC47. It is likely that H2S mediates sensory nerve excitation by hypoxia by inhibiting mitochondrial ETC, thereby affecting K+ conductance of type I cells (Figure 1).
Studies with genetically engineered mice suggest that the inactivation of NDUFS2 is an important step for the type I cell response to hypoxia. However, it remains to be determined whether graded hypoxia inhibits NDUFS2 and establishes the affinity of O2 for this enzyme. NDUFS2 is a ubiquitously expressed enzyme in the body. However, unlike many other tissues, CBs are highly sensitive to changes in O2 levels. Consequently, the uniqueness of NDUFS2 signaling in the CB remains to be established.
Finally, whether HO-2 and NDUFS2 signaling operates independently or works in concert is not clear. The CB responds to a wide range of pO2 values (about 80–20 mmHg). It was proposed that interactions between multiple signaling pathways working in concert as a “chemosome” enable the CB to sense a wide range of pO2 values45,46. Given that high concentrations of H2S can inhibit mitochondrial ETC47, HO-2 and NDUFS2 signaling might work in concert as a “chemosome” for the full expression of the CB to a wide range of hypoxia, a possibility that remains to be investigated.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)