Keywords
Hidradenitis Suppurativa, Cytokeratin, Immunohistochemistry, Pathogenesis, Inflammation, Follicular Occlusion
Hidradenitis Suppurativa, Cytokeratin, Immunohistochemistry, Pathogenesis, Inflammation, Follicular Occlusion
Based upon the comments of reviewers the following changes have been made:
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Hidradenitis suppurativa (HS) is a chronic inflammatory disease, the exact pathophysiology of which remains incompletely defined1. Numerous inflammatory mediators including TNF-α2, IL-172,3, IL-324 and IL-36 subtypes5,6 have been implicated in the disease. However, there is an incomplete understanding of the source and triggers of these mediators and how they sustain the chronic inflammation that characterizes this disease1,2. The pathogenic paradigm of HS has evolved dramatically since the first description by Velpau in 18397. First thought of as an apocrinitis of infectious aetiology, it is now considered a disorder of follicular occlusion and more recently an inflammatory disease characterised by a keratinocyte mediated inflammatory response6. However, the variable response to topical, systemic and biologic therapies in HS8 indicate our understanding of disease pathophysiology is incomplete when compared to other cutaneous inflammatory diseases such as psoriasis9 and atopic dermatitis10. Existing studies examining the histology and immunohistochemical profiling of HS tissues represent conflicting results, for example in the degree of dermal dendritic cell infiltration11,12 and the production of TNF-alpha in the follicular unit13,14. These results may be influenced by heterogeneous sampling methods, laboratory processing methods and data analysis15. An additional complicating factor is that clinical comorbidities which are strongly associated with disease activity in HS, such as obesity16, diabetes17, inflammatory bowel disease18, and smoking19 also impact inflammatory cell activity in the skin18,20–22. Hence it remains unclear whether the presence or absence of these conditions may confound the findings of immunohistochemical studies in HS15,23 and whether clinical stratification of patients is required to identify distinct pathogenic pathways, which may be amenable to pharmacological intervention. This variability across studies makes comparing data problematic. To date no systematic analysis of immunohistochemical studies has been undertaken to compare results, methodology and analytical techniques.
The objectives of this systematic review are:
This systematic review was registered with PROSPERO24 (Registration number CRD42018104763) and was conducted in line with the PRISMA25. The STROBE statement26 was used to assess the observational studies included in this study.
Information Sources for this review encompassed Pubmed (1946-July 1 2018), Scopus (2004- July 1 2018) and Web of Science (1990-July 1 2018) as shown in Figure 1. Search strategy is presented as Table 1.
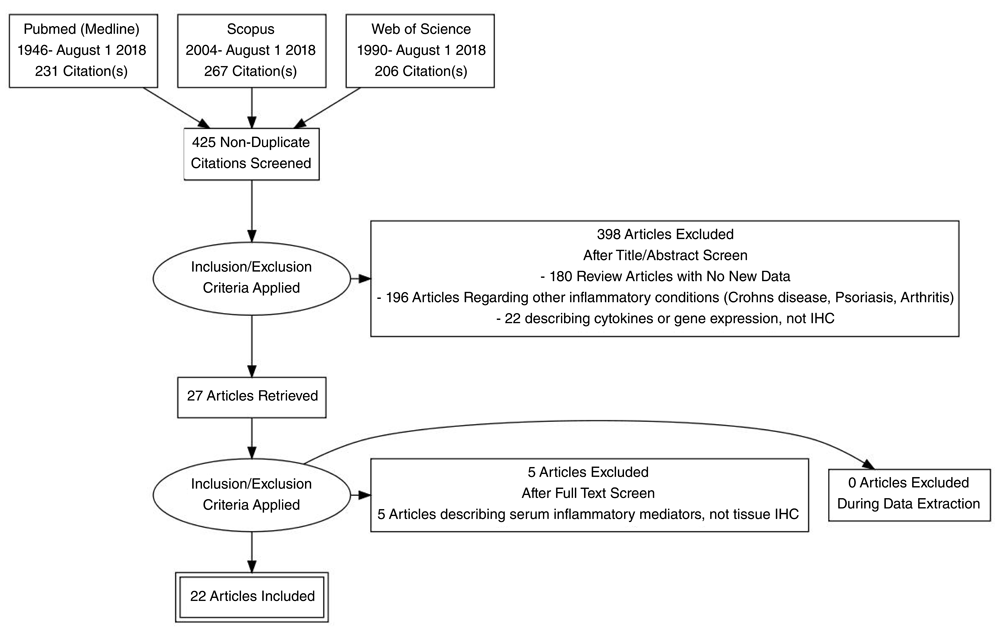
Eligibility criteria for this review included cohort studies, case-control studies and other observational studies with no restrictions of patient age, sex, ethnicity or language of publication. Eligible studies included those reporting the results of immunohistochemical findings in HS. Studies deemed not eligible included articles which provided no new data, only a review or summary of previously published data.
Data collection was performed independently by 2 authors (JWF & JEH), with any disagreements regarding inclusion of citations being referred to a third author (JGK) for mediation. Information was collected using a standardized data collection form (available as Extended data27) with the principal outcomes of interest being the immunohistochemical stain of interest, the site and rated intensity of staining (as described by authors), and comparison with perilesional/ unaffected/ control tissue. If data from individual patients was not available then the aggregate data was collected.
Potential sources of bias in the identified studies are acknowledged including the small size of patient cohorts, the variability in sampling and laboratory techniques, antibodies published and reactants used. Therefore these variables (where available) were collated to assess the heterogeneity of studies. Bias was also assessed using the NIH quality assessment tool for observational studies28.
A total of 425 non-duplicated citations were identified in the literature review (Figure 1). 398 of these articles were removed upon review of titles and abstracts against the pre-defined eligibility criteria. Full text review of the remaining 27 articles excluded 5 articles providing no new data. The remaining 22 studies4–6,11–14,29–43 reporting the results of 494 individual HS patients and 168 control patients were used as the basis of this systematic review.
The demographics of the patients of the included studies are presented in Table 2. Of 494 HS patients, 180 were male (38.3%) and 290 female (61.7%) with 24 cases unreported. Ages ranged from 15–72 years. 47/50 (94%) of reported cases were smokers, 12/30 (40%) had a BMI >30, and there was no information pertaining to diabetes or family history of HS. Of the 200 documented biopsy sites 93 were axillae (46.5%), 69 were inguinal (34.5%), and 38 were genital (19%) (Table 3). 64 patients had Hurley staging with 7/64 (10.9%) Stage 1, 40/64 (62.5%) Stage 2 and 17/64 (26.6%) Stage 3. No individual Sartorius scores were reported. Where current treatment was reported, 6 patients (4.5%) were on adalimumab, 42 (31.6%) were untreated, 85 patients (63.9%) had treatment withheld prior to biopsy, and 357 cases were unreported. Lesional biopsies were taken from all studies, with 3 individual studies also taking perilesional biopsies6,30,41. Age and Sex matched controls were present in 3 studies29,31,43 and results were stratified in a minority of studies. 2 studies stratified by disease severity4,12, 7 studies stratified by lesion site13,33–37,39, 5 studies stratified by treatment4,5,12,29,32, and no studies stratified by comorbidities. Analysis of immunohistochemical staining methodology varied and included quantitative analysis (3 studies)14,30,32, semi-quantitative analysis (14 studies)4,5,11–13,29,34,37–43, and the presence or absence of staining (5 studies)6,31,33,35,36. A total of 87 distinct immunohistochemical staining targets were identified (Table 4, Table 5 and Table 6).
| Number of HS Patients | Male | Female | Mean Age (Years) | Comorbidities | Biopsy Sites | Hurley Staging | mHSS Score (Mean) | Therapy | Study Reference | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Smoking | Obesity (BMI>30) | Diabetes | Family History | Axillae | Groin | Genital | ||||||||
| 18 | 11 | 7 | (Range 19–62) | NR | NR | NR | NR | NR | NR | NR | 14 | |||
| 15 | 6 | 9 | 38.7 | NR | NR | NR | NR | 9 | 4 | 2 | Stage 1=0 Stage 2=10 Stage 3=5 | NR | Nil | 6 |
| 24 | 8 | 16 | 36.5 (range 21–51) | NR | NR | NR | NR | NR | NR | NR | Mean=2.29 (SD=0.62) | NR | Untreated | 29 |
| 22 | 10 | 12 | 38.2 (Range 19–60) | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | 30 |
| 10 | 5 | 5 | 42 (Range 21–49) | NR | NR | NR | NR | Y | Y | N | Stage 2 (100%) | NR | Treatment Withheld | 32 |
| 20 | 8 | 12 | 37.5 (Range 21–51) | N=18 | N=10 | NR | NR | NR | NR | NR | NR | NR | Treatment Withheld (8 weeks) | 4 |
| 25 | 9 | 16 | 36 (Range 18–51) | NR | NR | NR | NR | NR | NR | NR | Mean =2.16 (SD=0.55) | NR | Treatment Withheld (3 weeks) | 5 |
| 47 | 19 | 28 | 42.3 (Range 22–54) | NR | NR | NR | NR | 48.3 (Range 8–144) | NR | 31 | ||||
| 11 | 9 | 2 | 39.6 (Range 18–61) | NR | NR | NR | NR | NR | NR | NR | “Mod-Severe Disease” | NR | NR | |
| 20 | 6 | 14 | 40 (SD=15) | 19 | 27.6 (4.1) | NR | NR | 7 | 12 | 1 | Stage 1=4 Stage 2=11 Stage 3=5 | NR | Treatment withheld 3 weeks prior | 12 |
| 10 | 1 | 9 | 38 (SD=15) | 10 | 28.9 (SD 4.5) | NR | NR | 3 | 7 | 0 | Stage1=2 Stage2=7 Stage3=1 | NR | Treatment Withheld 3 weeks prior | |
| 14 | 1 | 30 | NR | NR | NR | NR | 1 | 0 | NR | NR | NR | 13 | ||
| 1 | 42 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 25 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 22 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 45 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 27 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 38 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 34 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 59 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 41 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 33 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 46 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 49 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 1 | 31 | NR | NR | NR | NR | 1 | NR | NR | NR | |||||
| 60 | 26 | 34 | 37.3 (Range 15–67) | NR | NR | NR | NR | 1 | 6 | 1 | NR | NR | NR | 33 |
| 9 | 1 | 47 | NR | Y | NR | NR | 1 | 3 | NR | adalimumab | 11 | |||
| 1 | 31 | NR | N | NR | NR | 1 | 1 | NR | adalimumab | |||||
| 1 | 24 | NR | N | NR | NR | 1 | 3 | NR | adalimumab | |||||
| 1 | 32 | NR | N | NR | NR | 1 | 3 | NR | adalimumab | |||||
| 1 | 58 | NR | N | NR | NR | 1 | 3 | NR | adalimumab | |||||
| 1 | 58 | NR | N | NR | NR | 1 | 2 | NR | adalimumab | |||||
| 1 | 36 | NR | Y | NR | NR | 1 | 2 | NR | Nil | |||||
| 1 | 39 | NR | N | NR | NR | 1 | 3 | NR | Nil | |||||
| 1 | 67 | NR | N | NR | NR | 1 | 3 | NR | Nil | |||||
| 16 | 1 | 15 | NR | NR | N | NR | NR | 3 | 13 | NR | NR | NR | 34 | |
| 5 | 1 | 4 | 18-36 | NR | NR | NR | NR | 2 | 3 | NR | NR | NR | 35 | |
| 50 | 18 | 32 | 11-70 | NR | NR | NR | NR | 39 | 6 | 5 | NR | NR | NR | 36 |
| 14 | 11 | 3 | 16-72 | NR | NR | NR | NR | 2 | 12 | 0 | NR | NR | NR | 37 |
| 15 | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | 38 |
| 22 | 6 | 16 | 45.6 (Range 29–69) | NR | NR | NR | NR | 13 | 7 | 2 | NR | NR | NR | 39 |
| 9 | 3 | 6 | 44 (Range 32–70) | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | 40 |
| 12 | 0 | 12 | 29.4 (Range 19–42) | NR | NR | NR | NR | 3 | 0 | 9 | NR | NR | NR | 41 |
| 36 | 13 | 23 | 25 (Range 20–69) | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | 42 |
| 10 | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | 43 |
| 494 | 180 | 290 | 47/50 Reported | 12/30 Reported | None Reported | None Reported | 93/200 | 69/200 | 38/200 | Hurley 1= 7 Hurley 2= 40 Hurley 3= 17 Unknown= 430 | No Individual Scores Reported | adalimumab=6, untreated=42, treatment withheld=- 85, not reported= 357 | ||
| IHC Targets | Number of HS Patients | Number of Controls | Samples Analyzed | Age/Sex Matched Controls | Stratified by severity | Stratified by lesion site | Stratified by Co- morbidities | Stratified by Treatment | Immunostaining Intensity Assessment | Study Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| α-MSH, LL37, S100A7, MIF, TNF-α, hBD3, lysozyme | 18 | 12 | L | N | NR | N | N | N | Quantitiative Immunohistomorphometry (Image J Software) | 14 |
| IL36 | 15 | 15 | L, PL | NR | NR | N | N | N | Present/ Absent | 6 |
| CD3, CD56 LL37 | 24 | 9 | L | Y | NR | NR | N | Y (untreated) | Semiquantitative (0-3) | 29 |
| CD1a, CD4, CD8 CD20, CD56, Factor XIIIa, IL17, NLRP3, Caspase-1 | 22 | Yes (NR) | L, PL, U, C | NR | N | N | N | N | Cell Counting square grid x400 magnification | 30 |
| IL-23, IL-12, CD68, CD4 | 10 | 8 | L, C | N | N | N | N | Y (ceased 3/52 prior) | Positive stained cells per mm2 | 32 |
| IL-32 | 20 | 10 | L, C, S | N | Y | N | N | Y (ceased 8/52 prior) | Semiquantitative (+ to ++++) | 4 |
| IL-36 | 25 | 7 | L, C, S | N | N | N | N | Y (ceased 3/25 prior) | Semiquantitative (+ to ++++) | 5 |
| LCN2 | 10 | 16 | L | Y | N | N | N | N | Present/ Absent | 31 |
| CD11c | 20 | 6 | L | N | Y | N | N | Y | Semiquantitative (+ to ++++) | 12 |
| MMP2 hBD2 TNF-α | 14 | 2 | L, C | N | N | Y | N | N | Semiquantitative ((+ to ++++) | 13 |
| CD3, CD4, CD8, CD68 CD79 CD56 | 60 | Yes (NR) | L,C | N | N | Y | N | N | Present or Absent | 33 |
| CD3, CD4, CD8, CD20, CD138, CD14, CD68, CD11c | 9 | Yes (NR) | L,C | N | N | N | N | N | Semiquantitative (+ to ++++) | 11 |
| GCDFP-15, CD15, Lysosyme, S100, Ca19-9, HMB45 | 13 | 3 | L,C | N | N | Y | N | N | Semiquantitative (+ to ++++) | 34 |
| CD29, CTx-FITC | 5 | 4 | L,C | N | N | Y | N | N | Present or Absent | 35 |
| AE1/AE3/PKC26/ Enhanced Alkaline Phosphatase | 50 | Y (NR) | L,C | N | N | Y | N | N | Present or Absent | 36 |
| K1, K10, K14, K16,k17, K19, | 14 | 1 | L,C | N | N | Y | N | N | Semiquantitative (+ to ++++) | 37 |
| Desmoplakin 1,2, Plakoglobin, Plakophilin 1,2, Desmoglein 1,2,3, Desmocollin 1,2,3,K2e, K4, K5, K6, K7, CK8,CK9, CK10, K13, K13/15/16, K14, K17,K19, K20 Ki67 | 15 | Y (NR) | L,C | N | N | N | N | N | Semiquantitative (+ to ++++) | 38 |
| ER, AR | 22 | 10 | L,C | N | N | Y | N | N | Semiquantitative (+ to ++++) | 39 |
| TLR2, CD3, CD19, CD56,CD68, CD11c, CD1a, CD206, CD207, CD209 | 9 | Y (NR) | L,C | N | N | N | N | N | Semiquantitative (+ to ++++) | 40 |
| TLR 2,3,4,7,9,ICAM-1, TNF-α, IL-6, IL-10, TGF-β, α-MSH, hBD2, hBD4 IGF-1 | 12 | Y (NR) | L,PL | N | N | N | N | N | Semiquantitative (+ to ++++) Epidermis Only | 41 |
| hBD3, S100A7, RNase7 | 36 | 57 | L,C | N | N | N | N | N | Semiquantitative (+ to ++++) | 42 |
| MMP8 | 10 | 8 | L,C | Y | N | N | N | N | Semiquantitative (+ to ++++) | 43 |
| 494 | 168 | 3/22 | 2/22 | 7/22 | 0/22 | 5/22 |
Table 3: Critical Evaluation of Methodology of Studies Included in This Review Key:L= Lesional, PL= Perilesional, U= Uninvolved, C= Control S=Serum, Y=Yes, N=No, NR= Not Reported, CTx-FITC =Cholera Toxin
| Cell Type | Study | Results |
|---|---|---|
| Basal Keratinocytes | 13 | MMP2 Expressed |
| Suprabasal Keratinocytes | 31 | LCN 2 staining of suprabasal keratinocuytes |
| Dermal Fibroblasts | 13 | MMP2 expressed |
| 7 | 33/51 specimens associated with ++ fibrosis | |
| Neutrophils | 13 | MMP2 in keratinocytes, fibroblasts, macrophages and lymphocytes, |
| 30 | Significant increase in number of neutrophils in dermis Dermis> Perifollicular | |
| 31 | LCN2 in neutrophils – epidermis and dermis | |
| 7 | +++ infiltrate in 29/51 specimens | |
| Plasma Cells | 7 | +++ Plasma cell infiltrate in 2/51 specimens |
| Eosinophils | 7 | +++ Eosinophilic infiltrate ++ in 3/51 specimens |
| Histiocytes | 7 | +++ Infiltrate 24/51 speciments |
| Lymphocytes (NOS) | 13 | MMP2 expressed TNF alpha positivity in dermis |
| 44 | Lymphocytes, giant cell and necrosis in established lesions | |
| T Cells | 4 | CD3 + Dermis producing IL32C D 56 + NK T cells producing IL32 |
| 33 | lymphocytic mixed infiltrate perifollicular (with unruptured terminal follicles). This consisted of CD-3 (39%), CD-4 (30%), CD-8 (14%), and positive cells (CD-4 ⁄ CD-8 ratio: 2.1:1). CD-56 (0.1%) and UCHL-1 (0%) brought no conclusive results. Conspicuous was a CD-8 cell positive folliculotropism in all immuno- histologies (Figure 3). CD-8 positive lymphocytes were loosely distributed not only in the stratum basale but also in the suprabasal epithelial areas. The subepidermal inflammatory infiltrate in the area of interfollicular epidermal hyperplasia showed a comparable cellular composition: CD-3 (38%), CD-4 (26%), CD-8 (19%), CD-56 (0.2%) and UCHL-1 (0%), CD-4 ⁄ CD-8 Ratio: 1.4:1. Here too, a CD-8 positive pronounced epidermotropism was impressive | |
| 30 | At perifollicular sites, quantitative analysis showed a significant increase in the mean number of CD3+, CD4+ and CD8+ T lymphocytes (CD3+, 34 ` 20 per HPF; CD4+, 38 ` 21; CD8+, 12 ` 8) compared with healthy control skin (CD3+, 9`4; CD4+, 2`1; CD8+, 1`1; | |
| 3 | CD4 T cells producing IL17 in dermis | |
| 32 | CD4 T cells producing IL17 in dermis | |
| B Cells | 11,12 | Psuedolymphomatous nests (see cytokine studies) |
| 33 | Perifollicular infiltrate with unruptured terminal follicles: CD-79 (35%) Subepidermal interfollicular Infiltrate: CD-79 (33%), | |
| Dendritic Cells | 11 | Successful Adalimumab treatment reduced influx of CD11c+ dendritic cells in lesional skin |
| 12 | Number of dendritic cells stable in skin- mild elevation only | |
| 4 | Dermis producing IL32 | |
| Macrophages | 13 | MMP2 expressed TNF alpha positivity |
| 30 | Significant increase in deep infiltrate | |
| 32 | Increase with co-staining of CD68/CD32 and IL12/ IL23 | |
| 33 | Perifollicular infiltrate with unruptured terminal follicles: CD-68 (12%) Subepidermal interfollicular Infiltrate: CD-68 (19%), | |
| 4 | Dermis producing IL32 | |
| Mast Cells | 30 | Significant increase in deep infiltrate |
| 12 | Significant increase in deep infiltrate |
| IHC Staining Target | Epidermis | Dermis | Hair Follicles | Sinus Tracts | Subcutis | Apocrine/ Eccrine Glands | Study Reference | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Suprabasal Staining | Basal Staining | Dermal Staining | Infundibular Staining | ORS Staining | Type 1 | Type 2 | Type 3 | |||||
| Type A | Type B | Type C | ||||||||||
| CD1a | + | 30 | ||||||||||
| ++ | + | 40 | ||||||||||
| +++ | 12 | |||||||||||
| CD3 | ++ | ++ | ++ | 30 | ||||||||
| + | + | + | 40 | |||||||||
| 33 | ||||||||||||
| CD4 | ++ | 12 | ||||||||||
| Interfollicular and perifollicular | 33 | |||||||||||
| + | + | 30 | ||||||||||
| 32 | ||||||||||||
| CD8 | + | 12 | ||||||||||
| Epidermotropism | Interfollicular and perifollicular | 33 | ||||||||||
| + | + | 30 | ||||||||||
| CD11c | +++ | 12 | ||||||||||
| +++ | 40 | |||||||||||
| CD14 | 12 | |||||||||||
| CD15 | + | 34 | ||||||||||
| CD19 | - | + | 40 | |||||||||
| CD20 | +++ | 30 | ||||||||||
| +++ | 12 | |||||||||||
| CD29 | + | + | + | + (NOS) | 35 | |||||||
| CD32 | 32 | |||||||||||
| CD56 | 30 | |||||||||||
| - | + | 40 | ||||||||||
| CD68 | Deep> Perifollicular | ++ | + | 30 | ||||||||
| Interfollicular and perifollicular | 33 | |||||||||||
| + | +++ | 40 | ||||||||||
| 32 | ||||||||||||
| CD79 | Interfollicular and perifollicular | 33 | ||||||||||
| CD138 | Mild infiltrate | 12 | ||||||||||
| CD206 | + | +++ | 40 | |||||||||
| CD207 | +++* | + | 40 | |||||||||
| CD209 | ++ | ++++ | 40 | |||||||||
| Cytokeratins | ||||||||||||
| AE1 | Single K | 36 | ||||||||||
| AE3 | Single K | 36 | ||||||||||
| PKC26 | Single K | 36 | ||||||||||
| Factor XIIIa | DC + | 12 | ||||||||||
| + | + | 30 | ||||||||||
| K1 | Present in acanthotic epidermis | + | - | + | + | - | - | 37 | ||||
| K2e | ++ | - | - | - | 38 | |||||||
| K4 | - | - | - | - | 38 | |||||||
| K5 | + | + | + | + | 38 | |||||||
| K6 | - | + | + | + | 38 | |||||||
| K5/6§ | ++++ | 30 | ||||||||||
| K7 | - | - | - | + | 7 | |||||||
| K8 | - | - | - | - | 38 | |||||||
| K9 | - | - | - | - | 38 | |||||||
| K10 | - | 36 | ||||||||||
| Present in acanthotic Epidermis | + | - | + | + | - | - | 37 | |||||
| ++ | + | ++ | - | - | 38 | |||||||
| K13 | - | + | + | - | 38 | |||||||
| K13+15+16§ | + | + | + | + | + | 38 | ||||||
| K14 | Highly positive in acanthotic epidermis | + | + | + | + | ++ | Sebaceous Duct and Gland + | 37 | ||||
| + | + | + | 38 | |||||||||
| K15 | Stained apocrine glands | 34 | ||||||||||
| K16 | Weakly positive in acanthotic epidermis | - | + | - | + | + | - | 37 | ||||
| K17 | Weakly positive in acanthotic epidermis | - | + | - | + | + | - | 37 | ||||
| K18 | 38 | |||||||||||
| K19 | + NOS | + | 36 | |||||||||
| Weakly positive in acanthotic epidermis | - | + | - | - | - | - | 37 | |||||
| - | - | ++ | 38 | |||||||||
| K20 | - | - | - | 38 | ||||||||
| Ki67 | + | ++ | ++ | ++ | 38 | |||||||
| ER | - | + | 39 | |||||||||
| AR | - | NC | 39 | |||||||||
| GCDFP-15 | Apocrine glands | 34 | ||||||||||
| S100 | Eccrine glands | 34 | ||||||||||
| Lysozyme | Vulval cases only | 34 | ||||||||||
| ↓ in scarred cases | 14 | |||||||||||
| HMB45 | - | Negative all cases | 34 | |||||||||
| TLR2 | ++ | ++++ | 40 | |||||||||
| ↓ | 41 | |||||||||||
| TLR3 | ↓ | 41 | ||||||||||
| TLR4 | ↓ | 41 | ||||||||||
| TLR7 | ↓ | 41 | ||||||||||
| TLR9 | ↓ | 41 | ||||||||||
| ICAM-1 | ↓ | 41 | ||||||||||
| TGF-β | ↓ | 41 | ||||||||||
| IGF-1 | ↓ | 41 | ||||||||||
| RNase7 | +++ | 42 | ||||||||||
| MMP2 | +++/++++ | +++/++++ | + | +++ | + NOS | 13 | ||||||
| MMP8 | (Neutrophils) | +++ | (Neutrophils) | 43 | ||||||||
| Cholera Toxin | Slopes of papillae suprabasal epidermis, | hair follicles | + NOS | 35 | ||||||||
| Desmoplakin 1 | ++ | ++ | ++ | + | 38 | |||||||
| Desmoplakin 2 | ++ | ++ | ++ | + | 38 | |||||||
| Plakoglobin | ++ | ++ | ++ | + | 38 | |||||||
| Plakophilin 1 | ++ | ++ | ++ | + | 38 | |||||||
| Plakophilin 2 | - | - | - | 38 | ||||||||
| Desmoglein 1 | ++ | + | ++ | ++ | - | 38 | ||||||
| Desmoglein 2 | + | + | ++ | ++ | 38 | |||||||
| Desmoglein 3 | ++ | ++ | ++ | + | 38 | |||||||
| Desmocollin 1 | ++ | + | ++ | - | - | 38 | ||||||
| Desmocollin 2 | ++ | + | ++ | ++ | + | 38 | ||||||
| Desmocollin 3 | ++ | ++ | ++ | + | 38 | |||||||
| hBD2 | ↓ | Negative in 12/14 | 1 | |||||||||
| ↓ | 41 | |||||||||||
| hBD3 | ++ (suprabasal) | - | ++ | 14 | ||||||||
| +++ | + | 42 | ||||||||||
| hBD4 | ↓ | 41 | ||||||||||
| TNF-α | ++/+++ (macrophage/ lymphocytes) | ++/+++ | +++ | 13 | ||||||||
| ++ | ++ | + | NC | ↓ | 14 | |||||||
| IL-6 | ↓ | 41 | ||||||||||
| IL-10 | ↓ | 41 | ||||||||||
| IL-12 | ++++ | 32 | ||||||||||
| IL-23 | ++++ | 32 | ||||||||||
| IL-17 | Diffuse | 30 | ||||||||||
| +++ | 32 | |||||||||||
| IL-32 | ++ | +++ | 4 | |||||||||
| IL-36 | + | +++ | 6 | |||||||||
| Suprabasal | +++ | 5 | ||||||||||
| Caspase1 | ++ | 30 | ||||||||||
| NLRP3 | ++ | 30 | ||||||||||
| MIF | ++ | ++ | 14 | |||||||||
| S100A7 | ++ | +++ | ++ | 14 | ||||||||
| ++ | 41 | |||||||||||
| LL-37 | ++ | ++ | NC | 14 | ||||||||
| ++ | +++ | ++ | 29 | |||||||||
| α -MSH | ++ | NC | 14 | |||||||||
| Target | Details | Study Reference |
|---|---|---|
| CD1a | CloneO10; Dako Cytomartion | 30 |
| CloneO10; Dako Cytomartion | 40 | |
| O10 1:20 Immmunotech, Prague, Czech Republic | 12 | |
| CD3 | clone F7.2.38; Dako) | 30 |
| Polyclonal rabbit anti-human CD-3 dilution 1:25; Dako Cytomation Denmark A ⁄ S, Glostrup, Denmark), | 33 | |
| Polyclonal 1:150 Dako, Glostrup, Denmark | 12 | |
| Clone PC3/188A; DakoCytomation, Glostrup, Denmark | 40 | |
| CD4 | 4B12 1:160 Monosan Uden The Netherlands | 12 |
| monoclonal mouse anti-human CD-4 dilution 1:10; Vision Biosystems Novocastra, Newcastle, UK | 33 | |
| clone 4B12; Dako | 30 | |
| MT310 Dako | 32 | |
| CD8 | C9/144B 1:100 Dako | 12 |
| monoclonal mouse anti-human CD-8 dilution 1:50; Dako Cytomation Denmark A ⁄ S | 33 | |
| clone C8/144B; Dako | 30 | |
| CD11c | 5D11 1:60 Novocastra Newcastle Upon Tyne, UK | 12 |
| Clone KB90 DakoCytomation | 40 | |
| CD14 | MY4 1:100 Novocastra Newcastle Upon Tyne, UK | 12 |
| CD15 | Not Reported | 34 |
| CD19 | Clone HD37; DakoCytomation | 40 |
| CD20 | clone L26 (1,4); Dako | 30 |
| L 26 1:400 Dako | 12 | |
| CD29 | fluorescein-tagged B-subunit of cholera toxin (CTx-FITC) + CyChrome (Pharmingen BD Biosciences, Franklin Lakes, NJ, USA) | 35 |
| CD32 | KB61 Dako | 32 |
| CD56 | clone 123C3; Dako | 30 |
| Clone MOC-1; DakoCyotmation | 40 | |
| monoclonal mouse anti-human CD-56 1:50; Dako Cytomation Denmark A ⁄ S), | 33 | |
| 123C3.D5 1:25 Thermo Fisher Scientific Altrincham UK | 12 | |
| CD68 | Clone PG- M1; Dako) | 30 |
| monoclonal mouse anti-human CD-68 dilution 1:50; Dako Cytomation Denmark A ⁄ S) | 33 | |
| Clone EBM11; Dako Cytomation | 40 | |
| KP1 1:160 Dako | 12 | |
| EBM11 Dako | 32 | |
| CD79a | monoclonal mouse anti-human CD-79 dilution 1:25; Dako Cytomation Denmark A ⁄ S) | 33 |
| JCB117 1:100 Dako | 12 | |
| CD138 | B-A38 1:25 IQ Products Groningen, The Netherlands | 12 |
| CD206 | Clone 19.2; BD Biosciences Pharmingen | 40 |
| CD207 | Clone DCGM4; Immunotech, Marseilles, France | 40 |
| CD209 | Clone DCN46; BD Biosciences Pharmingen, San Diego Ca USA | 40 |
| Cytokeratins | ||
| Pankeratin | AE1/AE3/PKC26; Ventana Medical Systems SA, Illkirch, Cedex, France | 36 |
| AE1/AE3 1:200 Thermo Fisher Scientific | 12 | |
| Factor XIIIa | AC-1A1 1:200 Thermo Fisher Scientific | 12 |
| clone E980.1; Leica Biosystems Newcastle, Newcastle upon Tyne, U.K.) | 30 | |
| K1 | 34 Beta B4 Novo Castra Laboratories Ltd, Newcastle- upon-Tyne, UK) | 37 |
| K2e | Ks2́ 342́ 7.1 against CK 2e (Dr L.Langbein, Heidelberg, Germany), | 38 |
| K4 | 6B10 against CK 4, | 38 |
| K5 | AE 14 against CK 5, | 38 |
| K6 | Ks6.KA12 against CK 6, | 38 |
| K5/6 | clone M7237; Dako | 30 |
| CK7 | OV-TL 12/30 and Ks7 ́18 against CK 7, | 38 |
| K8 | CAM 5 ́2 against CK 8, | 38 |
| K9 | HK9TY1 (guinea-pig polyclonal) against CK 9 (Dr L.Langbein) | 38 |
| K10 | Not Reported | 36 |
| LHP1 Novo Castra Laboratories Ltd, Newcastle- upon-Tyne, UK) | 37 | |
| MoAbs K8́ 60 and DE-K10 against CK 10, | 38 | |
| K13 | Ks13 ́1 against CK 13, | 38 |
| K13+15+16 | Ks8 ́12 against CK 13 15 16, | 38 |
| K14 | LL001 Novo Castra Laboratories Ltd, Newcastle- upon-Tyne, UK) | 37 |
| LL001 against CK 14, | 38 | |
| K15 | Not Reported | 34 |
| K16 | LL025 Novo Castra Laboratories Ltd, Newcastle- upon-Tyne, UK) | 37 |
| K17 | E3 Novo Castra Laboratories Ltd, Newcastle- upon-Tyne, UK) | 37 |
| Ks17.E3 against CK 17, | 38 | |
| K19 | Not reported | 36 |
| B170 Novo Castra Laboratories Ltd, Newcastle- upon-Tyne, UK) | 37 | |
| Ks19́1 against CK 19, | 38 | |
| K20 | IT-Ks20́10 against CK 20 | 38 |
| Ki67 | MIB 1 against Ki-67 | 38 |
| MIB1 1:100 Dako | 12 | |
| ER | ER (Thermo Scientific; pretreatment EDTA, pH 9.0, dilution 1:80). | 39 |
| AR | AR (Santa Cruz; pretreatment citrate, pH 6.0, dilution 1:100) | 39 |
| GCDFP-15 | Not Reported | 34 |
| S100 | Not Reported | 34 |
| Lysozyme | Not Reported | 34 |
| A0099 pAbG 1:100 rabbit antihuman Dako Corporation | 14 | |
| HMB45 | Not Reported | 34 |
| TLR2 | Clone TL2.3; Alexis Corp. San Diego Ca USA | 40 |
| Santa Cruz Biotechnology, Inc, Santa Cruz, California | 41 | |
| TLR3 | Santa Cruz Biotechnology, Inc, Santa Cruz, California | 41 |
| TLR4 | Santa Cruz Biotechnology, Inc, Santa Cruz, California | 41 |
| TLR7 | Santa Cruz Biotechnology, Inc, Santa Cruz, California | 41 |
| TLR9 | Santa Cruz Biotechnology, Inc, Santa Cruz, California | 41 |
| ICAM-1 | Beckman Coulter, Inc, Brea, California | 41 |
| TGF-β | AbD Serotec | 41 |
| IGF-1 | R&D Systems, Inc, Lille, France | 41 |
| RNase7 | Dako | 42 |
| MMP2 | MMP-2 (cat no. AF902, LOT DUB034081, obtained from goat, 1:100 dilution, R&D Systems | 13 |
| MMP8 | Dako | 43 |
| Cholera Toxin | fluorescein-tagged B-subunit of cholera toxin (CTx-FITC) + CyChrome (Pharmingen BD Biosciences, Franklin Lakes, NJ, USA) | 35 |
| Desmoplakin 1 | DP 1 2±2́15 and DP 1±2́17 against DP I II, | 38 |
| Desmoplakin 2 | DP 1 2±2́15 and DP 1±2́17 against DP I II, | 38 |
| Plakoglobin | PG 5́1 and PG 11E4 (Dr M.J.Wheelock, Toledo, OH, U.S.A.) against PG, | 38 |
| Plakophilin 1 | PP1-9E7 and PP1-5C2 against PP 1, | 38 |
| Plakophilin 2 | PP2- 150 against PP 2, | 38 |
| Desmoglein 1 | Dsg1E-P124 and Dsg1E-P23 against Dsg1, | 38 |
| Desmoglein 2 | Dsg2E-G129 and Dsg2E- G96 against Dsg2, | 38 |
| Desmoglein 3 | Dsg3-G194 and 5G11 against Dsg3, | 38 |
| Desmocollin 1 | Dsc1-U100 against Dsc1, | 38 |
| Desmocollin 2 | DC-Rab 36 (rabbit polyclonal) against Dsc2, | 38 |
| Desmocollin 3 | MoAb Dsc3-U114 against Dsc3, | 38 |
| hBD2 | Human beta-defensin 2 (cat no. AF 2758, LOT VJU015051, obtained from goat, 1:100 dilution, R&D Systems, Germany) | 13 |
| Abcam, San Francisco, California | 41 | |
| hBD3 | 1:400 rabbit antihuman Donated by Prof Schroders Labor Kiel germany | 14 |
| 1:1000 rabbit ani-human PeproTech, Rocky Hill, N J | 42 | |
| hBD4 | Abcam, San Francisco, California | 41 |
| TNF-α | TNF-α (code ab 6671, obtained from rabbit, 1:100 dilution, Abcam, Cambridge, UK | 13 |
| 559071 mAbG 1:10 mouse antihuman R&D Systems | 14 | |
| IL-6 | AbD Serotec, Oxford, England | 41 |
| IL-10 | R&D Systems, Inc, Minneapolis, Minnesota | 41 |
| IL-12 | IL-12p7024945 R&D Systems | 32 |
| IL-23 | IL23p19 HLT2736 Biolegend | 32 |
| IL-17 | clone AF-317-NA; R&D Systems, Wiesbaden Germany | 30 |
| Polyclonal R& D Systems | 32 | |
| IL-32 | NBP-76684, Novus (Littleton, CO, U.S.A.) | 4 |
| IL-36 | rabbit polyclonal anti-IL-36a (C-terminal; ab180909), rabbit polyclonal anti- IL-36b (C-terminal; ab180890) and mouse monoclonal anti-IL-36c (ab156783; all from Abcam, Cambridge, U.K. | 6 |
| AF 1078,1099,2320,1275 RnD | 5 | |
| Caspase1 | clone 14F468; Imgenex/Novus Biologicals, Littleton, CO, U.S.A.) | 30 |
| NLRP3 | clone Ab17267; Abcam, Cambridge, U.K. | 30 |
| MIF | MAB289 mAbG 1:100 mouse antihuman | 14 |
| S100A7 | HL15-4 mAbG 1: 20,000 mouse antihuman Donated by Prof Schroders Labor Kiel germany | 14 |
| LL37/ Cathelicidin | Ab64892 pAbG 1:1000 rabbit antihuman Abcam | 14 |
| Rabbit anti-human LL-37 [Abcam, Cambridge, UK | 29 | |
| α-MSH | M0939 1:500 Rabbit Anithuman Sigma | 14 |
| PROGEN Biotechnik GmbH, Heidelberg, Germany | 41 | |
| Tryptase | AA1 1:800 Dako | 12 |
| clone AA1; Dako | 30 |
Epidermis. The epidermis of HS lesional tissue expressed the normal array of keratins (K) in the basal (K5, K14) and suprabasal (K1, K2e, K10) layers. K6, K16 and K17 staining were increased compared to healthy controls in the suprabasal epidermis in one study30, however, K6 and K17 staining was not increased in the epidermis (only in non-keratinized portions of sinus tracts) in a second study38. Where K6 and K17 were positive in suprabasal epidermis, K17 staining was more pronounced than K6 staining30. K19 was weakly positive in acanthotic epidermis37. Ki67 staining was elevated in basal and suprabasal epidermis. Normal staining patterns of desmoplakin, plakophilin and plakoglobin were seen38. Cells staining positive for CD1a, CD206, CD207 and CD209 were seen throughout the epidermis40. CD3, CD4, CD8 and to a lesser degree CD68 positive cells demonstrated epidermotropism in sites of epidermal acanthosis30,33. CD29 and cholera toxin (double positive) staining cells were seen on the slopes of papillae of the epidermis35. hBD2 (human beta defensin) staining was decreased throughout the epidermis in two studies13,41 whilst hBD3 staining was increased throughout the suprabasal epidermis14,42, however only significantly in Hurley Stage 1 and 2 patients (p=0.045)42. hBD4 was decreased in suprabasal epidermis compared to healthy controls (p=0.001)41. Contradictory findings were seen in toll like receptor (TLR) 2 staining with an increase in the epidermis co-localizing with dendritic cells and macrophages in one study40 but suppressed in a second study41. Levels of TLR3, TLR4, TLR7, TLR9, ICAM-1, TGF-Beta and IGF-1 were only assessed by one study and all were suppressed throughout the epidermis compared with controls41. RNAase7 was increased in expression compared to healthy controls (p<0.05)42. MMP2 was positively expressed in keratinocytes throughout the epidermis13 and MMP8 in neutrophils within the epidermis43. TNF-α was highly expressed in macrophages and lymphocytes present in the epidermis, particular in the basal layers13,14 and NLRP3, MIF, S100A7, LL37/Cathelicidin and α-MSH all positive in suprabasal keratinocytes30,41. IL-6 and IL-10 were reported as suppressed compared to healthy control skin41, however, IL-36 subtypes were highly expressed in epidermal keratinocytes (more suprabasal than basal)5,6 with IL-32 also positive in the stratum granulosum4.
Dermis. CD1a, CD11c, CD206, CD207, CD209 and Factor XIIIa positive cells were identified in the dermis in three separate studies12,30,40, however the degree of infiltration varied. Dermal infiltrates of CD3, CD4, and CD8 positive cells, continuous with the epidermal infiltrates were a consistent feature of lesional HS dermis and were increased over controls30,33. The distribution of these cells was most pronounced in the interfollicular dermis (ie. towards the papillary slopes) and perifollicularly (ie. peri-infundibularly)30,33. CD56, CD68 and CD138 positive cells were diffusely seen throughout the dermis40. CD19 and CD20 positive pseudolymphoid follicles have been noted in other studies30,40. Single keratinocytes have also been identified in the dermis which stain with pancytokeratin markers (AE1/AE3/PKC26)36. Inflammatory cells in the dermis co-localized with TNF-α13,14, LL-37/cathelicidin29, IL-1232, IL-2332, IL-1730,32, IL-324, TLR240 and MMP843. MMP2 co-localized with macrophages and fibroblasts13. IL-36 was not identified in the dermis5,6.
Hair follicle. Cytokeratin staining of the follicular apparatus is consistent with normal K14, K16 and K17 staining. CD29 positive cells were identified in the infundibulum35. CD3, CD4, CD8, CD68, Factor XIIIa positive cells were seen within the outer root sheath (ORS) contiguous with dense peri-follicular inflammation in the adjacent dermis30,33. The presence of inflammatory cells co-localized with MMP213, TNF- α13,14, and LL37/cathelcidicin13,29. hBD313,42 and MIF13 also stained positive in the ORS. One conflicting study reported no change in TNF-α staining of the follicular unit13.
Sinus tracts. Staining patterns differed between superficial keratinized sinus tracts and deeper, inflamed non-keratinized sinus tracts. Normal epidermal cytokeratin staining was seen in the keratinized superficial portion of sinus tracts including K1, K10, K1436–38. Ki67 was elevated and CD29 positive cells were also identified in sinus tracts35. Ki67 stained in both keratinized and non-keratinised portions of the sinus tract38. K19 staining was absent in keratinized portions of sinus tracts37. In deeper, inflamed, non-keratinized portions of the sinus tracts, K16, K17 and K19 were positive, with loss of K1, K10 and adhesions molecules including DG1 (desmoglein 1)and DSC1 (desmocollin 1)37,38. Apocrine gland nuclei stained weakly positive for estrogen receptor39 and androgen receptor39, and these results were reported as no different from control specimens39. Lysozyme staining of apocrine glands was seen in cases of vulval HS only34.
Immunohistochemistry methods. The list of antibodies used for IHC staining is presented in Table 6. Consistent antibodies were used for CD1a; CD20 and tryptase staining, whilst different antibodies were used for other staining targets. Antibodies used were not described in two studies34,36.
Assessment of Bias. The result of bias assessment using NIH criteria is presented in Table 7. All 22 articles clearly stated the research question of interest with well-defined study populations. The application of inclusion and exclusion criteria, or the calculation of sample size, or effect estimates were not described in any study. Exposures (ie. the presence of disease) were established and measured in all studies prior to the outcome measures (IHC staining) being assessed and the disease was established for such a time that a relationship between exposure and outcome would be identified if one existed. Different levels of exposure (severity of disease) was taken into account in only two studies4,12 and was consistently measured using Hurley staging across all studies. No articles accounted for all possible confounding variables such as obesity, diabetes, family history or smoking status (Table 3).
| Study Reference | 1. Was the research question or objective in this paper clearly stated? | 2. Was the study population clearly specified and defined? | 3. Was the participation rate of eligible persons at least 50%? | 4. Were all the subjects selected or recruited from the same or similar populations (including the same time period)? Were inclusion and exclusion criteria for being in the study prespecified and applied uniformly to all participants? | 5. Was a sample size justification, power description, or variance and effect estimates provided? | 6. For the analyses in this paper, were the exposure(s) of interest measured prior to the outcome(s) being measured? | 7. Was the timeframe sufficient so that one could reasonably expect to see an association between exposure and outcome if it existed? | 8. For exposures that can vary in amount or level, did the study examine different levels of the exposure as related to the outcome (e.g., categories of exposure, or exposure measured as continuous variable)? | 9. Were the exposure measures (independent variables) clearly defined, valid, reliable, and implemented consistently across all study participants? | 10. Was the exposure(s) assessed more than once over time? | 11. Were the outcome measures (dependent variables) clearly defined, valid, reliable, and implemented consistently across all study participants? | 12. Were the outcome assessors blinded to the exposure status of participants? | 13. Was loss to follow- up after baseline 20% or less? | 14. Were key potential confounding variables measured and adjusted statistically for their impact on the relationship between exposure(s) and outcome(s)? |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Emelianov et al.14 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Hessam et al.6 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Thomi et al.29 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Lima et al.30 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Schlapbach et al.32 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Thomi et al.4 | Y | Y | N/A | N | N | Y | Y | Y | Y | N | Y | NR | N/A | N |
| Thomi et al.5 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Wolk et al.31 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Van der Zee et al.12 | Y | Y | N/A | N | N | Y | Y | Y | Y | Y | Y | NR | N/A | N |
| Mozeika et al.13 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Von Laffert et al.33 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Van der Zee et al.11 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Heller et al.34 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Gniadecki et al.35 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Fismen et al.36 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Kurokawa et al.37 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Kurzen et al.38 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Buiner et al.39 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Hunger et al.40 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Derno et al.41 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Hofmann et al.42 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Tsaousi et al.43 | Y | Y | N/A | N | N | Y | Y | N | Y | N | Y | NR | N/A | N |
| Total | 22/22 | 22/22 | N/A | 0/22 | 0/22 | 22/22 | 22/22 | 2/22 | 22/22 | 1/22 | 22/22 | NR | N/A | 0/22 |
The overall quality of data in this systematic review was sub-optimal with poor correction for potential confounding factors with only two of the 22 studies using objective measurement systems for IHC staining intensity4,12. The proportion of smokers was elevated (94%) compared to the rates of smoking in the HS population at large (70–89%)45. A number of studies (17/22) did not stratify results by treatment therefore there is a risk that staining intensity of pro-inflammatory mediators may be reduced due to concomitant treatment at the time of biopsy. The use of de-paraffinized tissue in retrospective studies30,33,34 can lead to false negatives in IHC dependent upon the preparation method of the original sample and the de-paraffinization process15. Hence there are factors in the population studied in this review which may bring into question the reliability of staining quantification. However, the presence or absence of IHC staining, particularly when confirmed in multiple studies is still considered reliable despite the risks of bias.
Conflicting results were identified in dermal CD1a staining12,30,40, dermal CK19 staining36–38, Epidermal TLR2 staining40,41 and TNF alpha staining in the follicular infundibulum13,14. Regarding CD1a staining, two of the studies reported only a mild dermal infiltrate of CD1a positive cells30,40, with a third study demonstrating a significant infiltration of these cells12. This third study clearly documented all treatment was withheld 3 weeks prior to the biopsies being taken12, whereas there is no description in the other two articles regarding the discontinuation or ongoing use of treatments30,40. Therefore, with the possibility of partially treated disease, an artificial reduction in the number of dermal dendritic cells is a possibility as treatment for HS (such as adalimumab) has been demonstrated to effectively reduce the infiltration of dendritic cells in vivo12. Similarly, studies examining TNF-alpha staining also differed in their stratification of patient based upon active treatment13,14. Significant reductions in TNF alpha staining were seen in the study with no documentation of treatment cessation14 when compared to the one study with clear documentation that all patients had treatment ceased prior to biopsy13. K19 staining was reported negative in all areas of the sinus tracts in one study37, whereas two additional studies36,38 described positive K19 staining in sinus tracts (one study non-specifically36 and the second in the deep inflamed, non-keratinized epithelium of the tract38). The difference between these staining patterns may be explained by the presence of inflammation. Kurzen et al.38 described the presence of K19 staining in non-keratinized epithelium of the deep sinus tracts only when associated with inflammation (Type 3 epithelia), staining was negative when no inflammation was present (Type 2 epithelia)38. Kurokawa et al. did not differentiate between inflamed and non-inflamed non-keratinized epithelium in their study37, and noted that the lesser degree of inflammation seen histologically may explain their differing results in comparison to Kurzen’s study37.
IHC staining, in particular co-staining with cellular markers and cytokines has enabled the localization of inflammatory mediators in order to ascertain the functional aspects of infiltrating inflammatory cells in HS, particularly highlighting the strong Th17 polarity of inflammation in HS3. A schematic representation of the pathogenesis of HS based upon the findings of this review is presented in Figure 2. This highlights the inter-relationship between inflammation and hyperkeratinization. Localization of TNF-α13, IL-1232, IL-2332 and IL-324, TLR240, MMP213, MMP843 and LL-37/cathelicidin14,29 production to infiltrating dermal macrophages and lymphocytes as well as localization of IL-36 subtypes5, LL-37/cathelicidin14,29, IL-1β32 and IL-2232 to keratinocytes illustrate the feed forward mechanisms similar to those seen in psoriasis9 and atopic dermatitis10 which likely contribute to persistent inflammation in HS. Rather than keratinocytes being innocent bystanders, these IHC findings demonstrate the central role keratinocytes play as producers of key inflammatory mediators as well as mediators of products (such as TGF-β and ICAM)41 that may contribute to fibroblast dysregulation and hypertrophic scarring46. A remaining unanswered question includes the temporal relationship between keratinocyte hyperproliferation and the activation of inflammatory cells infiltrating the dermis and epidermis in HS.
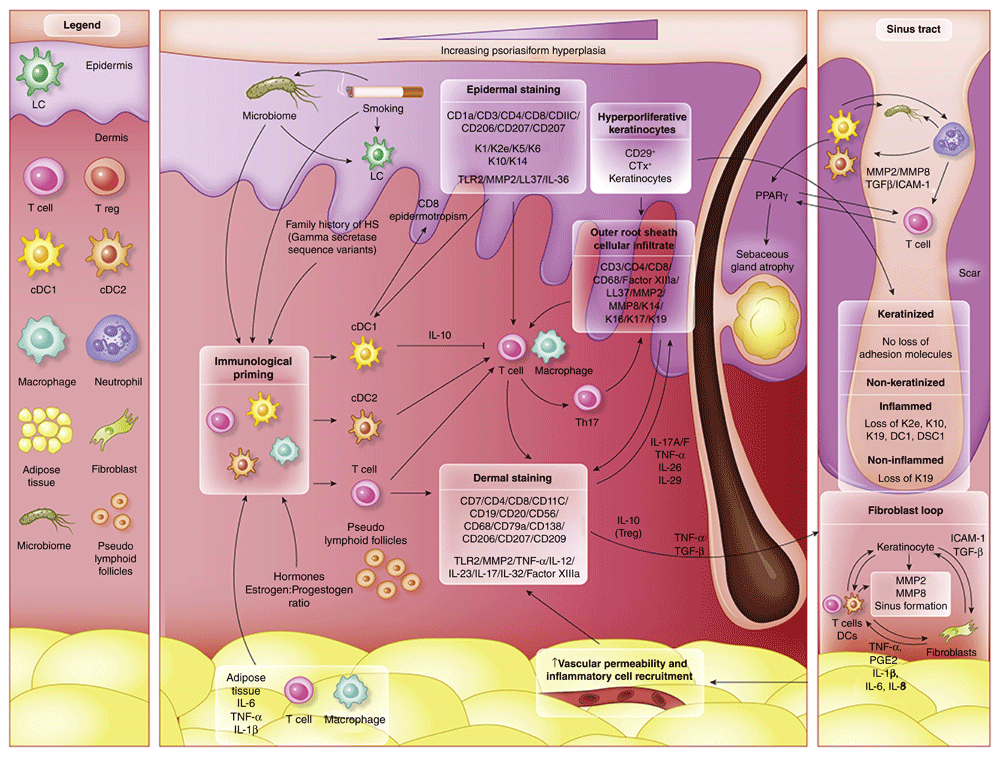
Immunological ‘priming’ occurs due to the contribution of adipose tissue, genetic susceptibility, smoking-related inflammatory mediators and obesity related pro-inflammatory signals and the composition of the microbiome. Increased activity of cDC1, cDC2 and T cells lead to both keratinocyte hyperplasia via the actions of IL-12 and IL-23, as well as a Th17 predominant immune response. Alterations of antimicrobial peptides (AMP’s) also occur throughout the epidermis. IHC staining localize Langerhan cells and activated dendritic cells to the epidermis and the dermo-epidermal junction. A population of epidermotropic CD8 T cells are also present. IHC staining indicates a mixed inflammatory infiltrate in the dermis, with contributions from Dendritic cells, B cells, T cells and plasma cells. Within sinus tracts, adhesion molecules are preserved, but inflammation in associated with non-keratinised sinus tracts leads to a loss of K19. The development of scarring and sinus tracts is associated with MMP2, ICAM-1 and TGF-Beta, with possible augmentation of ICAM-1 and TGF-B signaling via specific components of the microbiome. TNF-a, PGE2 and CXCL2 then lead to additional feed forward mechanisms perpetuating the inflammatory cycle.
The current pathophysiological paradigm of HS is one of follicular infundibular occlusion leading to follicle rupture and a resultant inflammatory cascade1. This paradigm was based on the pivotal work of Shelley and Cahn in 195547, whom demonstrated the induction of HS after application of belladonna impregnated tape to manually epilated axillae of 12 men. Only 3 of the 12 men developed the lesions described, and infection from the manual epilation procedure could not be excluded as a cause of the lesions, but this study enabled the paradigm to slowly shift away from one of apocrinitis, which had been in place since the original descriptions of the disease7. Detailed descriptions of infundibular hyperkeratosis (also termed poral occlusion) were made by Jemec et al.7 and demonstrated the secondary involvement of apocrinitis in HS lesions. Jemec noted that poral occlusion was seen to occur alongside inflammation, but there was no suggestion of causation in one direction or another7.
Although individual cases of epidermal hyperkeratosis in the absence of inflammation are noted7,33, these cases are established or chronic lesions associated with significant fibrosis which is documented to be associated with reduce inflammatory infiltrate7,33. A consistent finding in all studies of this review is the co-localization of infundibular ORS keratinocyte hyperplasia with CD3, CD4, CD8 and CD68 positive inflammatory cells expressing TNF-α, IL-12, IL-23 and IL-324,13,32,40,43. K19 is also documented as positive in the infundibulum suggesting keratinocyte hyperplasia36–38. However, it remains unclear whether keratinocyte hyperplasia induces the inflammatory cascade or if the inflammatory cascade induces the keratinocyte hyperplasia. The presence of inflammation in clinically normal, peri-lesional HS skin is well documented4,30,33 implying the existence of a pre-clinical inflammation preceding symptoms of follicular occlusion. This is consistent with recent findings in acne pathogenesis that suggest that inflammation precede follicular hyperkeratosis and development of microcomedones48 and is also pivotal in the ongoing development of nodulocystic acne and acne scars49. This pre-clinical inflammation is also consistent with the pathogenic paradigm in psoriasis and atopic dermatitis9,10 with inflammation driving epidermal hyperkeratosis and alterations in keratinocyte maturation, consistent with the spongiform infundibulfolliculitis seen in established lesions of HS50. Our disparate findings in K19 staining in deep non-keratinized sinus tract epithelia with and without inflammation37,38 also fit with this paradigm. In contrast, findings which would hold consistency with the current follicular occlusion paradigm would include infundibular occlusion preceding the development of inflammation, as well as alterations to desmosomal and hemidesmosomal proteins which would allow for rupture of the occluded follicles in order to drive the development of dermal inflammation and sinus tract formation. Although Danby et al.51 reports reduced PAS positivity in the basement membrane zone at the sebo-follicular junction associated with inflammation in HS, it is likely that the reduced basement membrane integrity is secondary to inflammation and release of TGF-β and MMP252 (cytokines known to be altered in HS lesional skin and consistent with an abnormal wound healing response) rather than the follicular rupture being the primary driver of inflammation.
A more consistent hypothesis which accounts for the observed results of this review would be that of subclinical inflammation (due to a variety of triggers and immunological primers as illustrated in Figure 2) driving keratinocyte proliferation in the interfollicular epidermis and the follicular ORS, with follicular occlusion being a secondary phenomenon (mediated by TLR2 and IL-1α as documented in the development of comedones)53. The development of sinus tracts and hypertrophic scarring may also be mediated by the keratinocyte inflammatory response given the alterations in important wound healing mediators including TGF-β, ICAM-1 and comparisons by other authors of an altered wound healing response8 in HS. This comparison would be appropriate given the high levels of dermal MMP213 and MMP843; the loss of keratinocyte maturation markers (K2e, K10, K19)36–38 adhesion molecules (DG1 and DCN2)38 in the non keratinized inflamed epithelium of the deep dermis; suppressed levels of ICAM-141 (seen impaired wound healing54) and TGF-β41 which leads to the dysregulation of TGF- β receptor ratio on fibroblasts which is linked with the development of hypertrophic scarring46,54 seen in HS. These alterations to keratinocyte maturation are reminiscent of epithelial mesenchymal transition (EMT)52 which may also explain the presence of free keratinocytes in the dermis in established lesions of HS7,36. Indeed, as ICAM-1 is up-regulated by pro-inflammatory mediators54, the low level of ICAM-1 noted appears paradoxical, however specific bacteria (including Porphyromonas species) which have been associated with HS44,55 can suppress ICAM-1 production as an immune evasion strategy56. This implies that exogenous triggers (possibly including bacterial stimuli) can be a common cause for the initial inflammatory cascade as well as the development of tunneling and hypertrophic scarring in HS.
This systematic review of immunohistochemical staining of lesions in HS has highlighted the heterogeneity of studies and the methodological issues, which bring into question some of the results of IHC staining in HS lesions. The design of studies and variable reporting of potential confounding factors (such as ongoing or previous treatments) makes it impossible to compare staining intensity across studies. The results of existing studies suggest a florid inflammatory reaction comprising of T-lymphocytes, macrophages and dendritic cells with a strong Th-17 signature along with a keratinocyte mediated IL-36 inflammatory loop associated with keratinocyte hyperproliferation. The follicular occlusion paradigm as a primary driver of HS is unclear given the findings of this review and other histological and cytokine studies and inflammation as a primary driver of disease with secondary hyperkeratosis and occlusion is a plausible hypothesis.
All data underlying the results are available as part of the article and no additional source data are required
OSF: Extended data. Data collection sheet. https://doi.org/10.17605/OSF.IO/2JKPW27
Licence: CC0 1.0 Universal
OSF: PRISMA Checklist for ‘A systematic review and critical evaluation of immunohistochemical associations in hidradenitis suppurativa’. https://doi.org/10.17605/OSF.IO/2JKPW27
Licence: CC0 1.0 Universal
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)