Introduction
Although Earl Benditt and his son John described the clonality of the atherosclerotic plaque in 19731 and although their observations have been confirmed many times2–7, interest in the idea that atherosclerosis might be a form of neoplasia waned because of the clinical success of treatments for hyperlipemia and because of the availability of transgenic mice as models for the effects of dysfunction of lipid metabolism on the vessel wall.
There are reasons to reconsider Benditt’s hypothesis. Over the last five years, the concept of clonality, both in cancer and in normal development, has changed. Over a longer period, we have also learned a great deal about the developmental biology of the normal vessel wall, including the normal formation of clonal patches in that wall and the development of a normal layer of smooth muscle cells (SMCs) in the intima long before atherosclerosis itself4,8. As a result, comparisons between atherosclerotic lesions and neoplasms now need to be rethought9.
Benditt’s hypothesis also needs to be revisited in terms of changes in how we define SMCs. We now know that these cells derive from several different mesodermal precursors, and recently criteria for defining cell types have been changing as a result of new insights driven by the analysis of single-cell expressomes.
Finally, since clonality of the lesions is real, the obvious questions are do these human tumors precede the development of atherosclerosis, how do the clones develop, what cell type gives rise to the clones, and in what ways do the clones provide the soil for development and natural history of atherosclerosis?
Does atherosclerosis begin with a neoplastic event?
Benditt’s 1973 paper10 was, to use a term familiar from today’s tech industry, “disruptive”. The paper showed that atherosclerotic lesions were clonal cell masses in the atherosclerotic intima. Benditt suggested that because clonality was a mark of cancer, the lesions arise as benign neoplasms11. This suggestion was based on an analogy to the benign uterine leiomyoma11–20. Like atherosclerotic plaques, uterine leiomyomas are monoclonal masses of SMCs. Like plaques, uterine leiomyomas are present in all humans, at least if “all” means most human females. Leiomyomas are made up of uterine smooth muscle. Using microdissection and immunocytochemistry, Benditt and Benditt identified the plaque clonal cell type with vascular SMCs. The team of Majesky, then a student working with Benditt Sr., went on to show that carcinogens, even without fat feeding, produced nodular smooth muscle tumors in the intima of chickens21 and so clonality of masses in the intima can arise as a neoplastic event.
Although Benditt’s data were repeatedly confirmed in human lesions2–7, interest in a “neoplasia hypothesis” was trumped. The success of lipid-lowering drugs maintained a focus on the traditional lipid hypothesis. The ease of studying atherosclerosis in experimental animals, especially inbred mice with transgenic modifications in lipid metabolism, permitted extensive studies of the arterial response to accumulation of lipid, even though the murine models have not, as of yet, fully modelled the advanced lesions that cause human death22,23.
Nonetheless, Benditt was at least formally correct. Spontaneous intimal masses (that is, smooth muscle-like collections in the intima) fit the usual definition of a benign tumor. In humans, these masses appear in the arterial intima prior to lipid accumulation24,25. The obvious questions are how do the intimal cell masses develop and in what ways do intimal masses provide the soil for development and natural history of atherosclerosis?
Figure 1 shows intimal cell masses in the arteries of human infants. These masses appear during development prior to lipid accumulation24,25. Evidence that these sites are clinically important came from the classic PDAY (Pathobiological Determinants of Atherosclerosis in Youth) study26. PDAY studied the arteries of young people who had died of non-cardiac causes. The most reproducible locations of advanced atherosclerotic lesions in these young people included the left descending coronary artery, the right coronary artery, the carotid bulb, and a region of the abdominal aorta below the diaphragm.
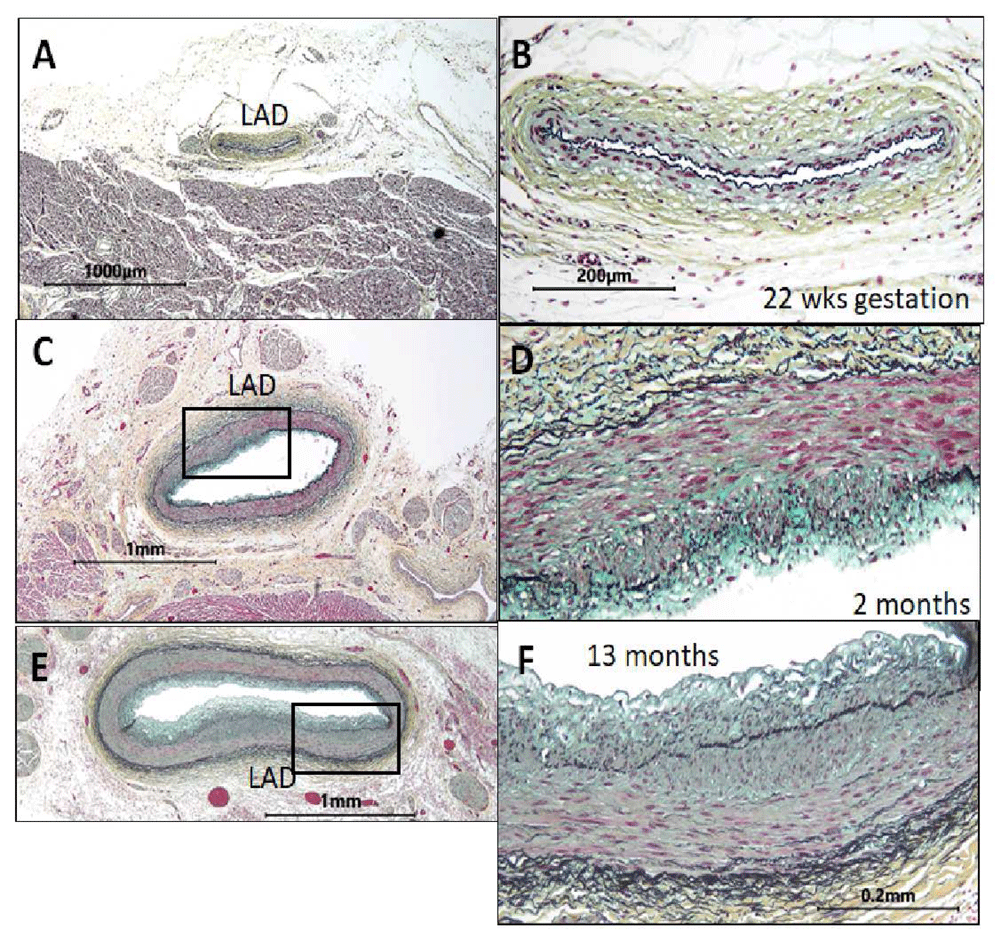
Figure 1. The normal intima occurs independently of and prior to the fat feeding associated with atherosclerosis4.
These images of the left anterior descending coronary artery (LAD) show the absence of cells in the intima in utero. The images to the right (B, D, and F) are enlargements of the LAD arteries to the right (A, C, and E). The endothelium at 22 weeks of gestation rests on the internal elastic lamina (A, B). By 2 months (C, D), a cellular intima is present in the LAD of this normal heart. By 13 months (E, F), this intima is about 50% as thick as the underlying media (400×, Verhoeff–van Gieson’s stain). In adult vessels, these normal sites would have progressed to form atherosclerotic plaques.
The sites identified in PDAY as being the most reproducible lesions are only a subset of the sites where fatty streaks, the initial lesions seen in fat-fed animals and in older human children, occur. Thus, atherosclerotic lesions can develop with and without pre-existing intimal cell masses. Moreover, there is no evidence that the development of intimal masses is necessary for the lipid-dependent processes leading to fatty streaks. Atherosclerotic diets fed to all mammals, including humans, result in accumulations of lipid- and fat-filled macrophages in the intima at arterial branch sites where blood flow is disturbed. Such sites include the openings of the intercostal arteries in fat-fed mice, rabbits, swine, or primates. Fatty streaks are also seen at branch points of thoracic arteries in young humans but recede as we age27,28. Further studies by DeBakey and Glaeser29 used angiography to follow the occurrence of newly formed lesions over 25 years in a large number of patients who initially had documented occlusive atherosclerosis at specific sites. The location of the new lesions in different arterial beds did not fit any well-described hypothesis such as distribution of blood flow. Perhaps the location of lesions was due to pre-existing intimal cell masses. Since these may be the sites where clinically significant lesions occur, perhaps the animal models do not fully model the human disease. Perhaps development of the fully developed, clinically significant lesions in adult humans depends on the initial processes, the cellular “soil” giving rise to a lesion in childhood.
Benditt versus Virchow
In Virchow’s seminal 1856 text, “Cellular pathology”30, the founder of modern pathology did not look at these earliest lesions. Instead, Virchow hypothesized that the adult atherosclerotic lesions he saw were the result of an inflammatory process stimulated by the toxic products of lipid accumulating in the intima. Migration and then proliferation of the cells we now call medial SMCs were part of Virchow’s view of this response to injury. Over 150 years later, Virchow’s “response to injury hypothesis”31 is supported by the ability of scientists to produce intimal lesions by feeding fat-rich diets to a variety of mammals, including mice, rabbits, pigs, and monkeys23,32–41. The “lipid hypothesis” or “response to injury hypothesis” remains dominant because of the obvious fact that lipid feeding in animal models produces lesions even in mice that lack a normal intima42,43 and the fact that lipid-lowering drugs greatly decrease the incidence of death from atherosclerosis in humans44.
It seems reasonable, based on studies in mice, to believe that medial SMCs are recruited into the intima to form a fibrous cap encapsulating the fatty accumulation45. Cell tracing studies now show that this fibrous cap in mice is derived from medial SMCs8,45–49. Moreover, recent studies show that this neointima, formed in response to the lipid-induced injury in mice, is clonal48,50. These cells surround lipid deposits and lipid-rich macrophages derived from blood monocytes formed much as proposed by Virchow. A huge amount has been learned about how the bone marrow-derived, fat-filled monocytes accumulate and enter the intima in response to inflammatory changes in endothelial cells induced by focal changes in blood flow or by hyperlipemia51.
If the mouse model is correct, fatty lesions begin before the formation of an intimal cell mass. If this were true in humans, clonality of atherosclerotic lesions might be irrelevant. However, we know that intimal cell masses develop at critical sites before lipid accumulation and, as we will discuss below, we still do not know how these cell masses form or why they become clones. Moreover, even the identity of the cells comprising the clones is unclear. Benditt’s use of “smooth muscle” to identify the intimal cells may itself have been simplistic because the identification of all cells that coat vessels as “smooth muscle” is being challenged. Recent reports show that some (or perhaps many) of these “plaque macrophages” are derived from medial SMCs that have assumed a macrophage phenotype49,52–55. Other studies raise issues about the use of smooth muscle alpha actin to define the SMC type. For example, recent studies show that the mural cells that form the coat around certain capillaries in the brain lack smooth muscle alpha actin56. Even if cells making up the vessel wall express smooth muscle markers (Table 1), cells comprising the mural cell coats have embryological origins from different regions of the mesoderm46,57–61. Thus, diverse “smooth muscle” subtypes that could give rise to the intima and diverse phenotypes could be important to the natural history and long-term outcome of the lesions.
Table 1. Mural cells, cell types that make up arterial wall coats around the endothelium.
| | Marker genes | References |
|---|
Medial smooth
muscle cells | Typical coats seen around endothelial and
endodermal tubes. Usually called “smooth
muscle” but, as discussed in the text, cells
lacking actin-rich contractile proteins have also
been seen in the media. | Smooth muscle alpha
actin, smooth muscle
myosin, smooth muscle
22α, calponin, and desmin | 62–110 |
| Intimal cells | Cells located between the endothelium and
the medial layers. Under endodermally derived
epithelium, these cells are called the “lamina
propria”. | See text. In general, intimal
cells have been described
as having markers shared
with smooth muscle cells. | |
| Neointimal cells | Cells that arise by migration from media or
adventitia into the intima. Neointimal cells may
also arise by transdifferentiation of endothelial
cells. | |
| Pericytes | Cells around small arteries and capillaries that
lack a medial layer | PDGFRβ, NG2, RGS5, and
smooth muscle alpha actin | 56,70,111–127 |
| Adventitial fibroblasts | Cells external to the media. These cells
typically lack smooth muscle markers but can
acquire them and become neointimal cells or
myofibroblasts. | FSP1, PDGFRα, periostin,
Tcf21, and cell surface
markers | 128–135 |
| Adventitial stem cells | Sca1, KLF4, CD34, Gli1,
and c-kit | 136–141 |
| Myofibroblasts | The major cells seen in fibrosis, at least in some
cases, arise from adventitial cells and express
smooth muscle proteins. | RGS5 and smooth muscle
alpha actin | 142–146 |
Normal intima versus neointima
As we have just discussed, intimal cell masses form spontaneously at specific sites in the intima of humans and other large animals4,43. We will refer to this cell mass as the “normal intima” or “normal cellular intima”. Other cellular intimas, called “neointimas”, form as pathological responses to almost any form of arterial injury42, including radiation, mechanical injury, immune responses, and, of course, accumulation of lipid in the atherosclerotic intima43.
Both neointima and, presumably, normal cellular intimas are prone to lipid accumulation when animals are fed a high-fat diet. Kovanen and Tabas have suggested that the extracellular matrix of cellular intimas may have properties that retain and accumulate lipid147. As already noted above, the recent observations that SMCs can differentiate into macrophages raise an entire new set of ideas about how intimal cells might predispose to lipid accumulation54,148. In summary, it seems likely that pre-existing cellular intima of spontaneous origin has properties that initiate atherosclerosis and perhaps determine the ultimate outcome of the lesions over the many decades of lesion progression in humans149.
Genetic versus epigenetic changes
Of course, neoplasia implies a genetic origin, a mutation as is believed to be the cause of uterine leiomyomas12,13,16,20,150–158. If uterine leiomyomas begin as mutations, why shouldn’t clonal masses in arteries also begin as mutations?
There are at least three ways Benditt’s clones might arise without the mutations implied by the neoplasia hypothesis. The first, as proposed at the end of this review, is isolation of some cells within the internal elastic lamina as the intima forms. The second is endothelial–mesenchymal transformation (EMT) as happens in cardiac valves159,160. The third is migration of rare cells from the media8,50,161. None of these hypotheses explains a selective advantage needed for clonal development.
The development of these initial rare cells into a clone suggests that some property causes the clone to survive and grow. The initial cell of the clone might grow because of properties of the milieu between the endothelium and the internal elastic lamella or because of unique properties of the subset of endothelial cells overlying the nascent lesion162. Alternatively, the clone might reflect epigenetic changes as have been reported for uterine leiomyoma163. Cells that make up the smooth muscle coats of arteries from diverse embryological origins8,58,164 may have epigenetic properties that confer genetic advantages. A relevant example of heritable changes associated with the origin of vascular smooth muscle may be the distinct properties of aortic cells derived from neural crest versus somitic mesoderm59,165–169.
Of mosaicism and clonality
As time passed and regardless of Benditt’s ideas about neoplasia, clonality in the vessel wall became well established. Twenty years after Benditt’s paper, Mikawa and Fischman used fluorescent viral tracing to show that the media of avian coronary arteries is formed by clones170. The fact that Mikawa and Fischman’s clones were spiral structures compared with the small number of cells comprising the arterial media of mice may explain the failure, so far, to identify large patch sizes (that is, clones) in the media of mice labeled by the confetti method2,3,5,9,46,171–176. Possibly, results will be different once bar code-tagged mice allow analysis of clonality as was done with zebrafish177.
Regardless of the findings in mice, we do know that the human media is made up of clones (that is, patches of cells that arise from single cells)178. Chung et al. were able to show that somatic mosaicism in the arterial wall was not limited to the plaque178. Mosaic patches present in the media led Chung et al. to propose that the plaque might arise by the amplification of cells from pre-existing patches in the underlying media178. As noted above, we now know that something like this happens in mice forming a neointima in response to injury3,46,50.
The idea that clonality is a normal part of tissue development is not unique to blood vessels. Highly accurate next-generation DNA sequencing is able to detect spontaneous mutations that occur as early as the blastula179. Such mutations can be used to trace development because we can cluster cells in terms of subsequent mutations179,180 (Figure 2). These methods permit the tracing of cell lineages of adult tissues as far back as events in gastrulation179. Although we know that somatic mutations are a normal part of development, most such mutations are not functional and are not causal for clonal expansion179,181. Somatic mosaicism is nonetheless intriguing. Clonal segregation of mutations in the brain may provide important mechanistic clues to the function of different regions in the brain182–184. Presumably, such mutations exist in the human vessel wall as well.
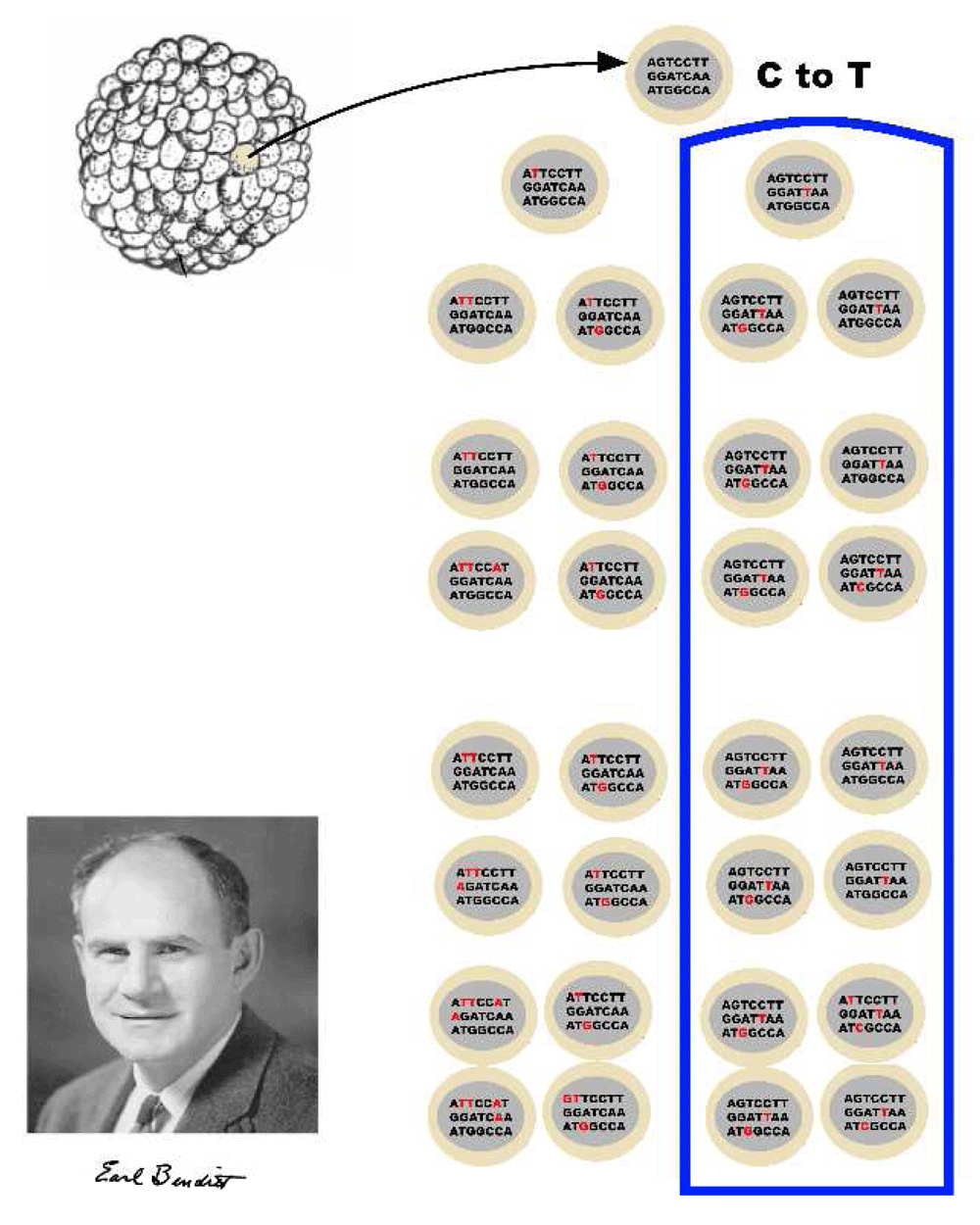
Figure 2. Clonality is normal.
Clonal origins of normal tissues detected by the identification of mutations using DNA sequence analysis. Benditt might be surprised to learn that he, like all humans, was a collection of clones!
The fact that plaques are clonal raises another possibility: functional mutations could arise in plaques as a result of, rather than as a cause of, clonal expansion. As a clone divides, replicating cells accumulate mutations that can provide a selective advantage over normal tissue, ultimately leading to a cancer181. Consistent with the idea that clonal expansion in humans leads to DNA damage, there are reports of mutations and DNA damage in the plaque171,185–216.
The diverse embryological origins of medial cells could provide a source of selective advantage for cells giving rise to an intimal clone. Embryological tracer studies of the media in mice have shown sources that range from mesectoderm derived from neural crest to mesoderm derived from the somites, the heart fields, mesothelium, and EMT8,49,59,128,164–166,169,217–229. In some parts of the arterial tree, multiple sources may be represented in a single part of the vessel wall. Perhaps medial cells derived from diverse origins have selective advantages for growth in the intima.
Finally, a recent study suggests that clonal expansion could be a cause of plaque rupture. Wang et al.149,230 demonstrated telomere shortening in cells of the plaque and suggested that the plaque cells forming the fibrous cap may, as a result of many replications, undergo cell senescence. They suggest that cell death in the cap eventually leads to plaque rupture149.
Clonality in the immune system
Surprisingly, a recent report shows that clonality of leukopoietic lineages in bone marrow is relevant to the final events in atherosclerosis195. Clonal diversity in the bone marrow decreases as we age while advantageous mutations undergo clonal selection231. Presumably, this is due to mutations in a highly replicative tissue combined with replicative senescence231. A mutation in the gene for the epigenetic modifier enzyme Tet2 promoted expansion of the mutant cells. One result of this is that leukemias in old age arise from a small number of surviving clones.
Fuster et al. showed that these changes in bone marrow lineages correlate highly with frequency of atherosclerotic clinical events195. The authors went on to show that the expansion of Tet2-mutant cells in atherosclerosis-prone mice accelerated lesion formation. They proposed that age-related increases in death from coronary artery disease reflect the effects of inflammation attributable to this mutation in plaque macrophages derived from the mutated myeloid precursors.
Because T- and B-cell clonal selection occurs early in life, plaque lymphocytes may not be affected by the Tet2 mutation. In any case, because Benditt showed that the clonal cell type in lesions was a smooth muscle, it is not likely that lymphocyte clonality accounts for plaque monoclonality. Moreover, studies by Hansson et al. of plaque T cells showed oligoclonality rather than monoclonality232.
“What is a ‘cell type’?”
As suggested above, the importance of clonality is tied to our concepts of cell type. In the 1850s, Robert Remak (Figure 3) observed red blood cells in chicken embryos. These cells differentiated from cell precursors through various stages of cell division. His concept, our modern idea of cell lineage, was put into Latin as “omnis cellula e cellula” (all cells come from cells) and became one of the central tenets of Virchow’s “Cellular pathology” text in 1856. Remak’s concept merged into the theory that cell type is determined by heritable changes that survive mitosis (Figure 4). The genetic concept of cell linage became central to the dogma that the origins of a metastasis could be defined by its resemblance to a normal tissue, an idea supported by the “unitarian hypothesis” of Maximov233,234. The idea became set in the modern biology canon in 2002 when Brenner, Horvitz, and Sulston won the Nobel Prize for tracing the full developmental lineage of all of the somatic cells, 959 for males and 1,031 in females, in the nematode Caenorhabditis elegans235.

Figure 3. Omnis cellula e cellula.
Robert Remak’s discovery of the modern concept of cell lineage is often falsely credited to Rudolph Virchow because Remak, as a Jew, had troubles getting published in mid-19th century Berlin237.
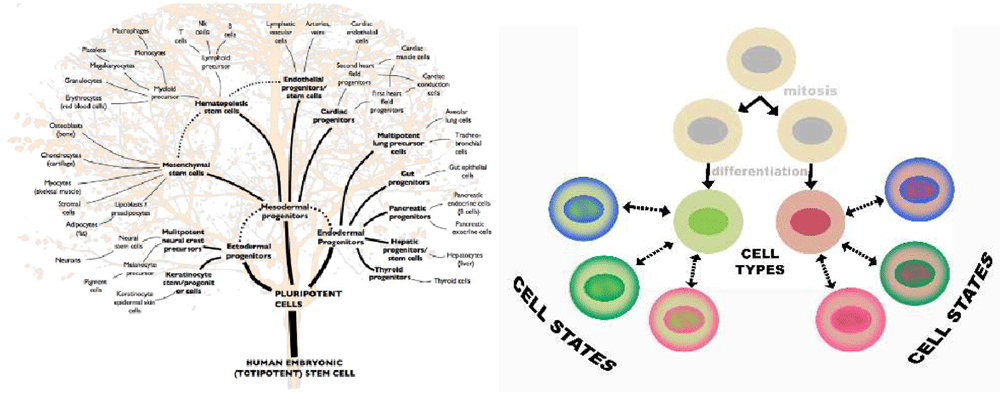
Figure 4. Cell type and cell phenotype are not necessarily the same thing.
Cells in a lineage can change phenotype in a process called “phenotypic modulation”. Recent data combining bar codes with single-cell RNA analysis show that cells can even transdifferentiate across traditional tree-like boundaries.
Over 150 years after the publication of “Cellular pathology”, Remak’s concept is now challenged because of a combination of new data in epigenetics, analyses of expressomes using single-cell RNA analysis, lineage analyses using spontaneous mutations or bar coding, and network analysis using Bayesian networks and cluster analysis236. We now know not only that cell types can branch in a tree-like fashion but also that, even independent of neoplasia, cells can jump between branches177 (Figure 5).
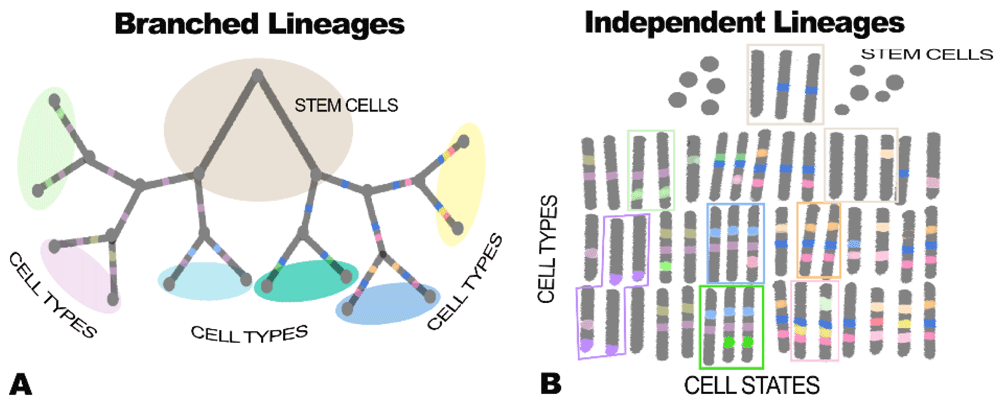
Figure 5.
Branched lineages (A) versus independent lineages with no branching (B). These two figures show very different ideas about how cells differentiate. In A, cells differentiate more or less the way species form (that is, in a branched tree arising genetically). In B cells, the model is very flexible, with very similar cell types, marked here by green and red rectangles, arising by the coordinated expression of different sets of genes under epigenetic control. Mathematical modeling using both marker genes and gene clusters shows that traditional branched tree (that, is branched lineage) cells can also switch phenotypes even between branches of the tree236. A formal way of thinking of this is that, rather than cell types, cells exist on the surface of a three-dimensional landscape as proposed by Waddington 60 years ago238. In this view, cells that are very much alike exist in valleys of coexpression as shown in part B of this figure and discussed by Trapnell239. Single-cell analysis of expression, combined with tools that can trace cell lineages in whole animals, now allows us to distinguish between branched and independent cell lineages as discussed in Figure 6.
It is important to understand that Remak and Virchow developed the ideas of cell lineage before Mendel. Darwinian evolution, from its onset, has been understood to reflect heritable changes in what we now call a genome. In contrast, as shown in Figure 5b, differentiation is not based on mutations in DNA and thus lineage maps do not need to follow a Darwinian set of branched pathways. For example, in macrophage differentiation, macrophages resident in different tissues retain an epigenetic memory of origin sites240, even when they move to new tissues. Moreover, macrophages themselves apparently can arise from very different lineages, including marrow-derived cells that originate from a subset of endothelium, tissue-resident macrophages that originate independently of the endothelium, and even SMCs46,148,162,240–247.
A new definition of cell type
The definition of cell types has traditionally been based on marker genes (that is, proteins found at high levels in a differentiated tissue). Examples include albumin for hepatocytes, immunoglobulins for B cells, insulin for pancreatic beta cells, vascular endothelial (VE) cadherin for endothelial cells, and cell type-specific muscle myosins or actins for heart and skeletal muscle. In the vascular media, cell type has been defined primarily by the location of cells and the high levels of expression of smooth muscle actin in cells organized as the tunica media (Table 1). Smooth muscle actin, however, is not definitive because not all cells in the tunica media express this protein248. Moreover, the expression of smooth muscle alpha actin is not restricted to smooth muscle. The gene is expressed in other muscle lineages as well as endothelial cells and even epithelial cells.
A new approach may be to define a cell type by transcription analysis based on sets of coregulated genes. These sets are called “clusters”249–251. Understanding how we can define cell types by “clustering” levels of RNA expression (Figure 6) requires that we consider how transcripts are measured. The accuracy of individual transcript levels may be less important than the numbers of transcripts measured in an assay. This is fortunate because, unless RNA levels are measured by quantitative polymerase chain reaction, efforts to determine the quantities of the 10,000 or more mRNA species in an individual cell type are fraught with error. These errors can be minimized in a number of ways, including using many probes for each putative RNA in a hybridization analysis, increasing the number of sequences identified by spending more money on sequence analysis, or clustering sets of coregulated genes250–253. Because there are many genes in a set, a mean value of expression for all genes in a cluster (that is, its center of gravity) has less error than the error of any one gene249–253. As a result, cluster analysis can be used to define cell types independent of any marker gene.
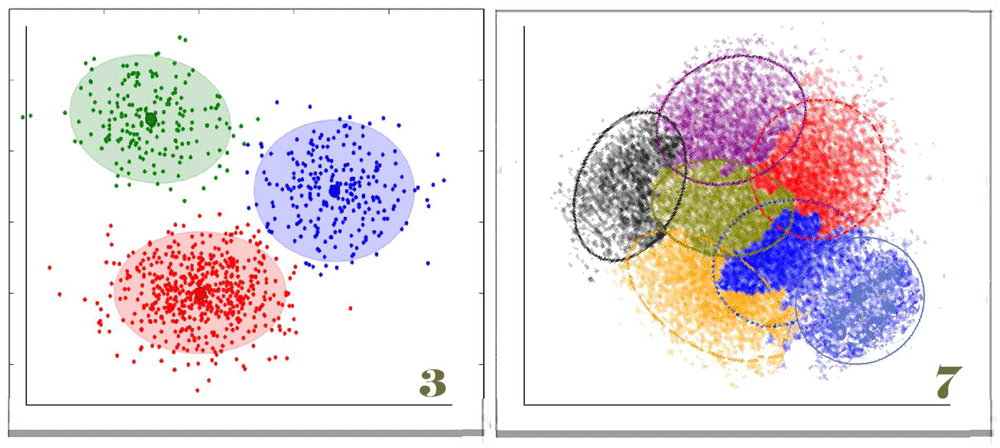
Figure 6. Defining cell type by cluster: three types and seven types?
Clustering cells by analysis of their mRNAs, or any other sort of single-cell data, is a way of arranging cells independently of assumptions about any structure other than similarity in different data sets. For example, clustering is independent of assumptions about lineage as shown in Figure 5A. If you have samples of mural cells from 100 aortas of different mice, you could ask how many cell types or groups of different cells make up the aortic media. Obviously, the clusters will include marker genes, but, because the number of genes sampled is large, the identity of each cluster as a “cell type” may or may not depend on that marker. Thus, a cell lacking the expression of a marker gene (for example, smooth muscle actin) might still be defined as a cell type based on the mean values of its other transcripts. Defining a cell type by cluster analysis has three major advantages over the use of marker genes. First, because the cluster is defined as the mean level of mRNA of the set of sampled genes, the noise level arising from measurement errors is much lower than a definition based on only a single value. Second, the cluster data can be combined with other data (for example, variations in genetic sequence) to determine pathways that control the cell’s phenotypes. Third, existing knowledge of expression pathways can be used to imply the mechanism providing overall control of the cell type. Cell type analysis defined by clustering can also be combined with lineage analysis using bar coding methods, analysis of the mutations that occur in the generations after zygosis (as in Figure 2), reporter genes that give cells different colors, or reporter genes that reflect the expression of particular genes at earlier stages of development. The result is that we can distinguish between cell types as defined by a cell’s ancestry and the cell’s phenotype at a specific time. The two cluster diagrams in this figure illustrate a need for caution. Even when, as on the left, the three clusters appear clear, the graphic image may reflect biases by the investigator. Clustering algorithms usually assume that the biologist wishes to see the number of clusters that offer the greatest separation between clusters. Programs either begin with a random guess about the number of clusters or use the number suggested by the investigators. The algorithms then move individual “genes” or other nodes between clusters to find an optimum that minimizes variability within clusters while maximizing variability between clusters. As shown in the diagram to the right with seven clusters, it is also possible that clusters overlap. Discussion based in part on http://www.statsoft.com/Textbook/Cluster-Analysis.
Of course, pathways are the ultimate definition of cell type and cell state. Covariation of the quantity of different RNAs (Figure 6) has been used for at least two decades to identify pathways. These pathways can be connected to causal Bayesian networks by identifying the sites of variation in gene sequence that control variation in expression254–258. These approaches have been used to define cell types in terms of a cluster of genes rather than individual marker genes259,260 (Figure 4–Figure 6).
Recently, in the vessel wall, cluster analysis has been used to define cell types using single-cell RNA analysis. Carried out over a time course and combined with the introduction of artificial DNA bar codes, this can be used to map the lineage of essentially every cell as an embryo develops261–263. The method has even been used to identify unexpected lineages when cloned stem cells were induced to develop into skeletal muscle239. Betsholtz and collaborators used cluster analysis to identify four different types of pericytes coating brain capillaries in the vessel wall56. Pericytes are smooth muscle actin-expressing cells that form the thin layer of cells surrounding the endothelium of capillaries (Table 1). One of these pericyte types, with a distinct location in the capillary bed, lacked smooth muscle alpha actin, the canonical marker for SMCs56.
Once defined, a cluster can be used to simplify transcription analysis of all the genes expressed in a cell. Moreover, since a few genes can be used to determine the expression of genes belonging to coregulated clusters253, accuracy of a cluster can be quite high. For example, hybridization chips that use fewer than 1,000 carefully curated probes to represent the entire transcriptome were recently designed253. These “L1000” chip probe sets reduce the cost of analysis of a transcript profile to less than $10. Because these chips are inexpensive and not limited by counting errors associated with RNA sequencing, very large numbers of cell types or cell states can be cheaply defined and stored in a common database. Currently, the consortium behind L1000 has analyzed 1.3 million samples representing different cell states or cell types253. The low cost of L1000 chips may make the identification of cell types and cell states very inexpensive. In summary, rather than single genes, clusters may provide a better way to define cell type56.
One concern is that the statistical construct of clusters may mean that we need to live with the idea that cell types overlap. Of course, that overlap may be the true meaning of cell type.
A semantic issue: “phenotypic modulation” versus “cell state”
A more general term than “phenotypic modulation” may be “change in cell state”. Whereas loss of cell differentiation in response to injury is common to most tissues responding to injury, the term “phenotypic modulation” is peculiar to vascular biology. The term had its origin in the early days of SMC culture when Julie and Gordon Campbell observed that cultured SMCs, like all other cells adapted to grow in culture, lost their differentiated phenotype. Cultured SMCs switched from a “contractile” in vivo to a “synthetic” phenotype adapted to growth in culture. The Campbells proposed that the loss of the contractile proteins, especially smooth muscle alpha actin, was central to the migration of medial cells and proliferation in the intima to form a neointima264,265. In subsequent work by Feil et al.62, Owens et al., and others46,53,62,63,266–275, much of the signaling process controlling this switch has been worked out. The signaling process involves transposition of a transcription factor from the promotor driving actin to a promoter driving genes required for cell proliferation.
As illustrated in Figure 4, changes in cell state occur in most cell types, including post-mitotic, terminally differentiated neurons that respond to a severed axon by switching to a phenotype that regenerates the injured axon276. As an extreme example, fibroblasts can be induced to differentiate into skeletal muscle by overexpression of the transcription factor myoD. The skeletal muscle type, however, is reversible if the cells are demethylated by introduction of bromodeoxyuridine (BUdR). When BUdR is removed, the cells redifferentiate presumably because of the persistence of epigenetic changes277. In this case, we might argue that the skeletal muscle cell type persisted even when the markers of skeletal muscle phenotype were gone.
The semantic issue may be whether one means “phenotypic modulation” to distinguish between reversible changes in phenotype (that is, cell states) or whether the term implies the more permanent properties that identify cell types. For example, most differentiated cell types can respond to tissue injury by adopting motile or replicative phenotypes. When the tissue has regenerated, the cells revert to their resting state. Even post-mitotic, terminally differentiated neurons are capable of undergoing chromatolysis. Chromatolysis is a massive increase in the proportion of DNA unfolded and involved in transcription as is needed to regenerate an injured axon276. Similarly, hepatocytes, skeletal muscle, renal tubular epithelium, and so on all undergo reversible changes in cell state when induced to regenerate after a wound.
As an example closer to vessel wall biology, the “myofibroblast” is derived from fibroblasts. The resting fibroblast lacks smooth muscle alpha actin or other markers of the smooth muscle type278. Following stimuli by certain cytokines, fibroblasts transform into a phenotype characteristic of a healing wound with an abundance of synthetic endoplasmic reticulum to make connective tissue and actin-rich microfibrils to promote wound closure. However, as the wound progresses, the myofibroblasts cease making matrix and lose their actin-rich content. They again appear as fibroblasts. Similarly, even if we define the clonal cells in a plaque as belonging to a smooth muscle “cell type”, it is not at all surprising that we can find SMCs in a proliferative or other state as required by the local environment46.
Whatever terminology we use, it is important to realize that Remak’s concept of cell lineage may no longer be correct (Figure 5). Wagner et al., combining single-cell RNA analysis with bar codes, found lineages that cross between the expected branches, implying that “cell types” can give rise to cells that otherwise would be assumed not to exist because of membership in a different lineage177. Wagner et al. concluded that “the ability of embryonic clones to undergo dramatic converging/diverging behaviors thus underscores a continued need for independent measurements of both cell state and lineage in the mapping of cell fate hierarchies”177. An important pathological example of a switch across lineages includes papers showing that glioblastoma stem cells, presumably of neural crest origin, are able to express both the endothelial phenotype and the SMC/pericyte phenotype279.
As already discussed, recent reports have described a change in smooth muscle phenotype that may even comprise a change in cell type. We already know that macrophages derived from cells in different organs retain epigenetic changes from that origin even when challenged with cytokines characteristic of different resident macrophage cell states243,244,280. However, it now appears that intimal cells that derive from medial smooth muscle can acquire macrophage properties52–55,148. The lineage issue becomes even more problematic with the recent reports that all macrophage and endothelial cells derive from a common precursor, the “erythro-myeloid progenitor” (EMP). In view of the ability of cells to cross traditional lineage barriers, it would be interesting to determine whether RNA expression data from these macrophage-like cells cluster with other tissue-derived macrophages, with macrophages derived from the bone marrow240,243,244,280, or perhaps even with endothelial cells undergoing endothelial–mesenchymal transformation.
In summary, we do not know whether phenotypic modulation allows an SMC to revert to a fully contractile phenotype. Put into conventional terms, perhaps intimal cells are their own “cell type”?
Origins and identity of “mural cells”: another semantic issue
The term “mural cell” is used in vascular biology for the connective tissue cells that form the wall around endothelial tubes. Because the term “mural cell” is based in morphology, the term can replace “vascular smooth muscle cell” without defining a cell type by lineage and without making assumptions based on the expression of a single gene (Table 1).
We need to understand how the formation of the endothelium determines the origins of mural cells (Figure 7). The angioblasts arise during gastrulation. Angioblasts are the progenitors of endothelium, monocytes, lymphocytes, and all white cells and red cells. Angioblasts, or perhaps the more primitive EMPs162, form endothelial cells as well as the precursors of tissue macrophages, including glial cells of the brain243. The endothelial cells form themselves into tubes, and these tubes join up and branch to form a capillary plexus. The plexus is a chaotic network that remodels into the orderly branched vascular tree with arteries, capillaries, and veins. Tissue macrophages, called histocytes, arise by migration of their precursors from the EMP. Monocytes arise from EMPs that become part of the endothelium and give rise to precursors that detach and populate the bone marrow and other hematopoietic sites.
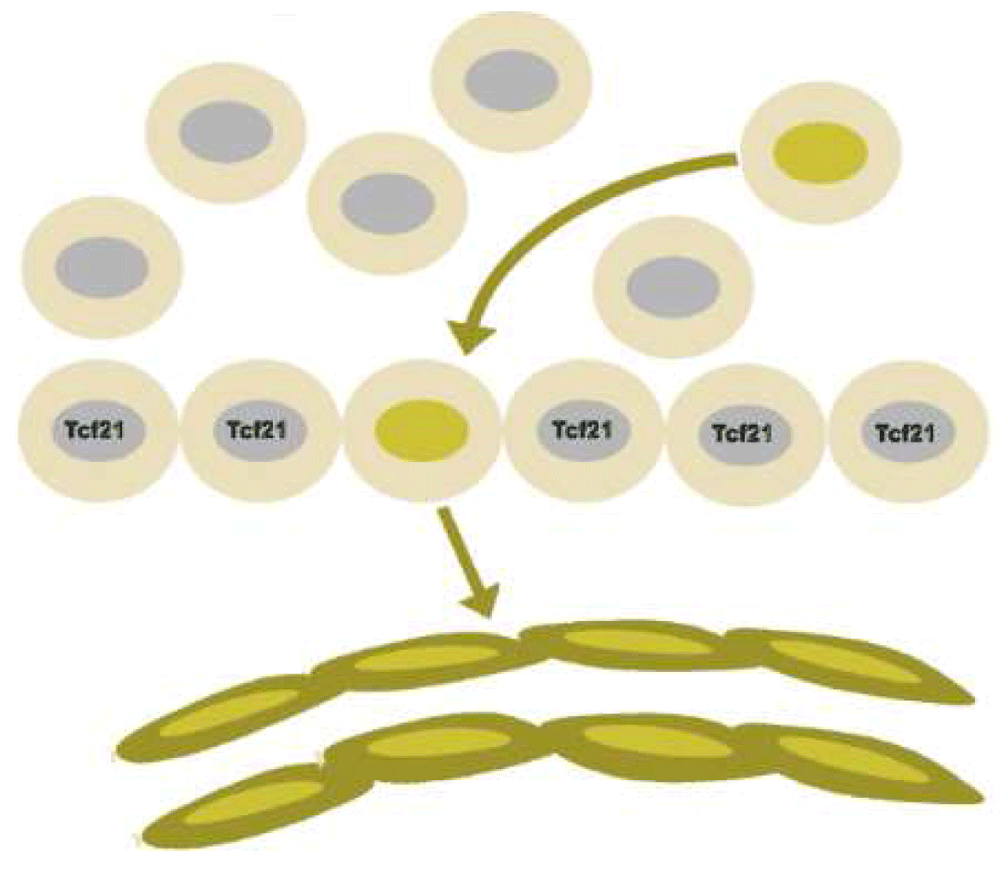
Figure 7. Endothelial cell precursors can be identified by lineage tracing within the proepicardial organ before cells from that precursor migrate to form the epicardium.
Cells marked by the expression of the transcription factor Tcf21, in contrast, give rise to the mesenchyme of the heart, including both smooth muscle cells and fibroblasts.
The branched endothelial tubes acquire coats of mural cells derived from various local forms of mesenchyme. The recruitment of mural cells does not occur until the branched tree of arteries and branches reaching all parts of the growing embryo have been formed. There is no evidence that marrow-derived cells give rise to precursors of mural cells.
Mesodermal sources of progenitors for mural cells are diverse. The sources range from the mesectoderm of the neural crest, the mesothelium of the thoracic and abdominal cavity, the heart fields, the endocardium, the epicardium, the metanephros, and the somites. In some places, mural cells may even develop by transdifferentiation of the endothelium49,59,166,222.
Not all mural cells go on to express smooth muscle alpha actin. In chickens, the media of the thoracic aorta contains two ultrastructurally distinct cell types, one cell having the characteristic filament-rich cytoplasm of an SMC and the other the ultrastructure of a fibroblast248. Embryological studies in chickens and mice further suggest that this morphology may represent unique origins from the somatic mesoderm versus the mesectoderm formed from neural crest. Similarly, mural cells surrounding certain capillaries in the brain fail to express smooth muscle markers56.
Given origins in distinct regions of the mesoderm, there is no reason to assume that the resulting cell populations would exist in a single-cell state, even if all of these cells were defined as being of one SMC type. Mural cells with different origins show distinct proliferative properties and biosynthetic profiles, even in cell culture64,169,281–285. In mice, Roostalu and Wong have recently shown distinct lineages within the media itself, including a distinct population of mural cells arising during embryonic development that is maintained postnatally at arterial branch sites226. Finally, similar studies show that multiple embryonic sources within the developing mouse heart contribute to the cells that become the medial smooth muscle of the coronary arteries286. As we will also discuss below, Tallquist et al. have shown that the epicardium gives rise to two distinct lineages: one that forms the media and another that forms the adventitia128.
Finally, data from studies of differentiation of organ-specific epithelium suggest that the origins of different mural cells might affect the cell state of organ-specific endothelial cells287,288. Differentiation of the endoderm into organ-specific types of epithelium depends on the interaction of epithelial cells with organ-specific mural cells and their matrix289–291. By analogy, it seems possible that different mural cells and intimal cells control the phenotype of overlying endothelium. Conversely, differences in lineage within the endothelium might control the formation of intimal cell masses162. Such cell type interaction-specific interactions between epithelium and mesenchyme might explain the localization of atherosclerosis at specific sites.
Possible sources other than mural cells for origins of intimal cells
The media is not the only possible source for intimal cells. Studies using labeled bone marrow cells in mice and humans have purported to show that intimal SMCs in atherosclerotic plaques could be derived from circulating marrow-derived cells292,293. These studies, however, have not held up to more careful work8,47,294.
A more likely source is the adventitia. The adventitia of arteries is defined as those cells outside the external elastic lamina (Figure 8). This layer contains endothelial cells, neurons, blood-derived macrophages, lymphocytes, mast cells, and fibroblasts. Recent studies show that adventitial fibroblasts include cells identified as stem-like by the expression of markers like c-kit and sca1224. Cell tracing studies show that adventitial cells can cross the media of injured vessels to form a neointima136,295,296. Moreover, transplantation studies in vivo show that adventitial stem cells applied to the outside of an injured vessel can migrate across the media and form an intima295.
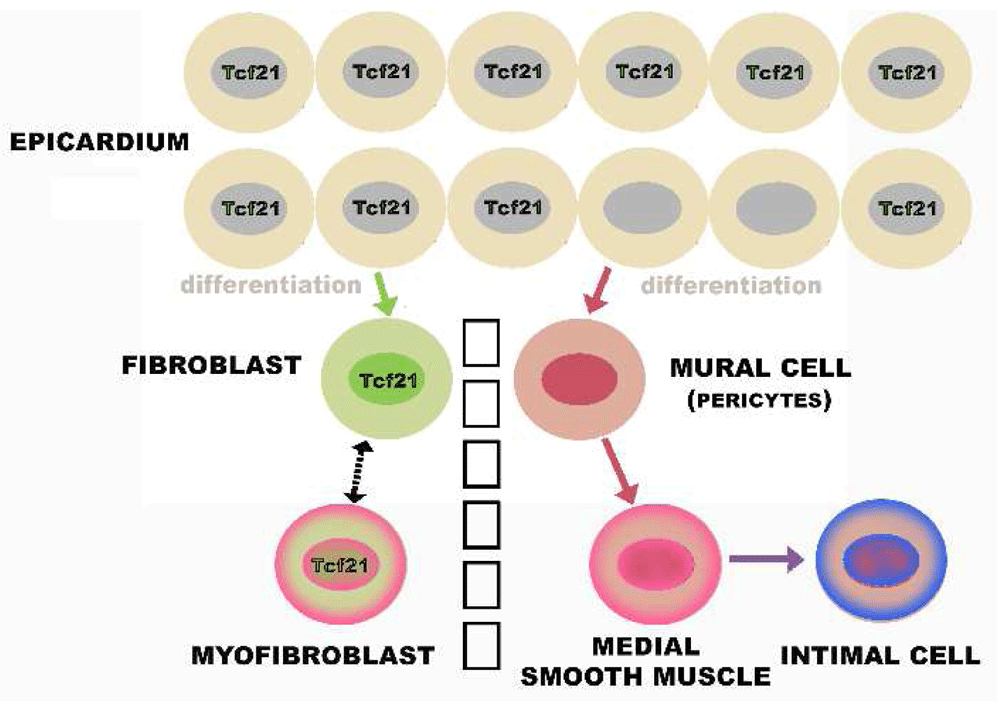
Figure 8. Layers of the artery wall.
The intima is a layer of connective tissue located between the endothelium and a layer of elastin called the internal elastic lamina. The media is delimited by the dashed black lines representing the internal elastic lamina and the external elastic lamina (EEL). Only rare cells, including lymphocytes and smooth muscle cells, are seen in the normal intima of the small mammals usually used to study atherosclerosis. However, in humans, intimal cells accumulate spontaneously during normal development and appear as a clone in the atherosclerotic lesions of adult humans. Based on immunocytochemistry, these cells are usually considered to be smooth muscle cells. In fat-fed animals, including humans, lipid accumulates in the intima to form the characteristic fatty atherosclerotic lesion. The outer limit of the tunica media is also defined by a layer of elastin called the EEL. Extrinsic to the EEL is a poorly defined tissue that is part of the matrix surrounding not just blood vessels but the parenchymal cells that comprise organs. The part of this matrix close to the vessel wall is called the adventitia.
Adventitial fibroblasts are also of interest because of their relationship to fibrotic responses. Outside the vessel wall, adventitial cells respond to injury by the synthesis of high levels of smooth muscle actin, becoming the major cell type seen in fibrosis, the “myofibroblast” described above297,298. Myofibroblasts characterize scleroderma and may be derived from vessel wall cells142. Myofibroblasts (that is, fibroblasts rich in smooth muscle actin) also characterize the mesenchyme of some tumors in a process called “desmoplasia”299–301. Curiously, no effort has been reported to use cluster analysis to compare myofibroblasts with intimal cells.
The origin of intimal cells from adventitial cells may imply that intimal cells are of a different cell type than medial cells. Tallquist et al. showed the adventitial cells of coronary arteries and the mural cells of the coronary artery media derive from distinctive precursors in the epicardium (Figure 9)128. This differentiation occurs when epicardial cells lose the expression of a transcription factor, Tcf21. The Tcf21-negative cells undergo epithelial–mesenchymal transformation, migrate to coat the nascent endothelial tubes, and form mural cells. These mural cells initially have the properties of pericytes and populate the entire coronary microvasculature302. In contrast, the fibroblasts around these vessels, that is the adventitial cells, originate from the Tcf21-positive epicardial cells by migration128. Presumably the adventitial cells express smooth muscle actin only during cardiac fibrosis when they become myofibroblasts. The observations of Tallquist et al. have not as yet been extended to other vascular beds128. Much less is known about the origin of adventitial cells other than those in the heart137,223,224,303.
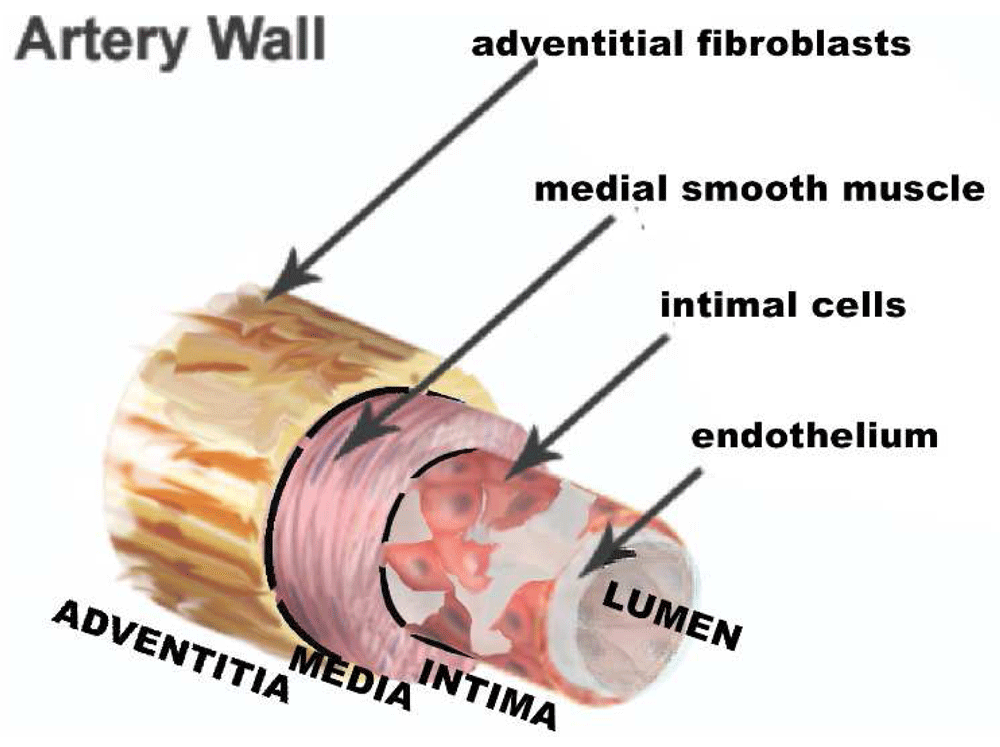
Figure 9. Dichotomous origins of medial smooth muscle and adventitial fibroblasts.
Both arise from the epicardium, but the smooth muscle lineage requires first the loss of expression of Tcf21. Presumably, the intimal cells arise from the medial smooth muscle but, as discussed in the text, we cannot rule out origin from the adventitia, especially since adventitial fibroblasts are the source of smooth muscle actin-rich myofibroblasts seen in injured myocardium.
There is, finally, one additional possible source for intimal cells, the endothelium. Endothelial cells are capable of undergoing EMT228,304–306. EMT has been intensively studied in the formation of the cells making up the cardiac valves129,160,304,307. In addition, Karsan et al. used the Tie1 promoter to identify an endothelial-like cell that is present in the vasculature of developing murine vessels. This labeled cell differentiates into smooth muscle. Endothelial cells that also showed the canonical VE cadherin marker, however, did not differentiate into smooth muscle308. It is intriguing to wonder if the normal intima might originate in such a peculiar precursor cell. The origin of the intimal cells of the human coronary artery from a human cell equivalent to the Tcf21-positive epicardial cells of Tallquist et al. or by EMT would provide an entirely new perspective on the meaning of clonality.
An intimal hypothesis based on fenestrae
Figure 10 proposes perhaps the simplest model for plaque monoclonality. In this model, rare mural cells are trapped within the intima as the internal elastica forms and the fenestrae, holes in the internal elastic lamina, shrink309,310.
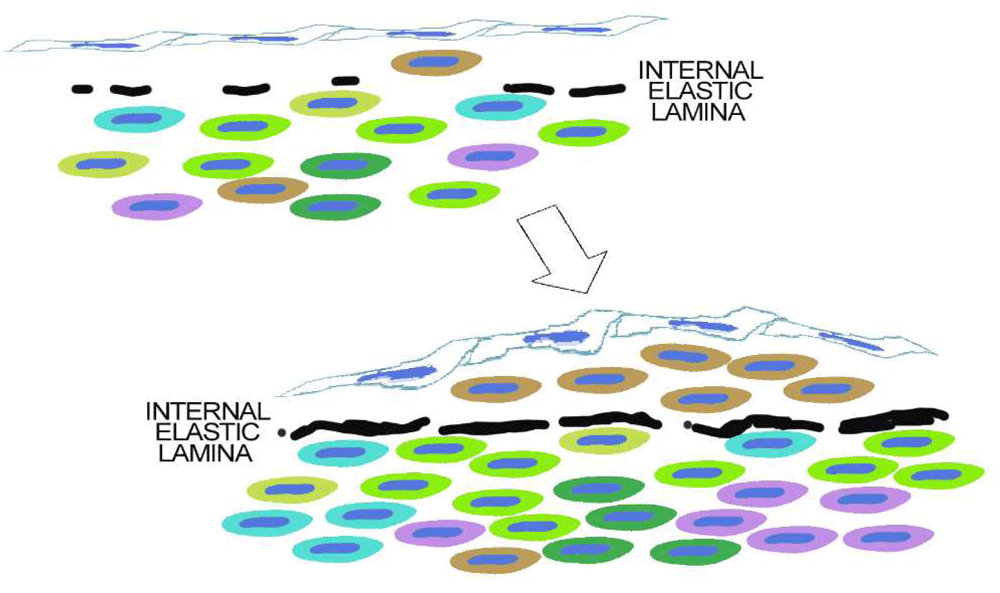
Figure 10. The simplest hypothesis.
As discussed in the text, intimal smooth muscle cells in adult human atherosclerosis are clonal. Although we do not know that this clone is derived from intimal clones present at birth, this is an obvious hypothesis. The next question might be how would normal intima develop as a clone? The simplest hypothesis is that some mural cells that coat the arterial endothelium get trapped within the forming internal elastica. Isolated from other mural cells, these intimal cells would develop a phenotype dependent on both their lineage and the conditions of being confined between the endothelium and the internal elastic lamina.
A simple alternative is that migration from the media is limited not only by the number and size of passageways across the internal elastic lamella but also by secretory and cytoskeletal changes required for transfenestral migration. Fetal properties that allow some medial cells to migrate may be developmental but could be reactivated in a subset of the medial SMCs when arterial injuries occur. A recent study by Lu et al.311 showed examples of transfenestral SMC migration in wild-type arteries after carotid ligation and in transgenic mice with Mef2c deletion specifically in endothelial cells.
Finally, localization of intimal cells might be the result of stimuli coming from endothelial cells responding to flow312 or from subsets of endothelial cells with their own lineages162.
Figure 10 does not rule out the possibility that the clone selected for growth in the intima has special properties derived from its developmental lineage, acquired because of the environment of an injured media, or resulting from mutation.
In the end, does intimal clonality matter?
We do not know whether the origin of a lesion in clones of intimal cells is important to the natural history of the plaques. The common belief is that atherosclerotic lesions develop selectively in areas where the arterial endothelium is exposed to turbulent blood flow312. However, as already discussed, we know that the neointimas formed after injury predispose to atherosclerosis and that sites with similar rheology may or may not develop atherosclerosis. Therefore, it seems likely that spontaneous and intimal masses may be the initial cause of atherosclerosis in humans.
Moreover, sites that show spontaneous intimal thickening26,313 are also sites that manifest in later life as lesions that rupture, leading to occlusion and thromboembolic events. Perhaps localization of lesions at these sites is accelerated because of synthesis of matrix proteins that bind lipoproteins314, because of pro-inflammatory interactions between endothelial cells and intimal cells51, because of regional differences in Hox gene expression and nuclear factor-kappa B (NF-κB) activity which correlate with areas of lesion formation in aortas of atherosclerosis-susceptible mice315, because of localized differences in the synthesis of elastin which eventually affect plaque stability316–240, because telomere shortening due to clonal amplification leads to senescence, cell death, and rupture of the fibrous cap149, or even because of immunological properties of the macrophage phenotype derived from medial SMCs53,320.
Summary
New frontiers that have emerged since Benditt’s original observation
Given all that we have learned from mice and from the success of lipid-lowering drugs, Benditt’s observation would be irrelevant today if we did not know, as in Figure 1, that the intimal cell mass precedes the development of the fatty lesions. Benditt’s hypothesis would still be irrelevant unless we imagined that the natural history of the plaque over decades of plaque development could depend on the identity and properties of the clonal cell type making up the intimal cell masses.
Our review has tried to make seven points:
1. Benditt’s observations have held up, but his neoplastic hypothesis has not. Even though carcinogens can promote the growth of intimal cell masses21, there is (as yet) no evidence that the clonal growth of the human plaque is due to a mutation. Perhaps such evidence will emerge with applications of next-generation sequencing to detect mutations in the plaque cells181.
2. Intimal cell masses in human arteries precede atherosclerosis. However, we do not know that these masses are clonal. It is conceivable that clonality develops later as lesions develop as is seen in mice responding to arterial injury.
3. Intimal cells may determine where plaques develop. It is possible that intimal cells control the phenotype of overlying endothelium, including properties that attract leukocytes leading to the development of atherosclerotic lesions. Intimal cells may also provide an environment that accumulates toxic products of lipids.
4. Intimal cells have properties distinct from our traditional view of SMCs. We do not know whether these properties are reversible or represent a fixed change (that is, something that might be considered a “cell type”).
5. Cluster analysis, combined with lineage tracking, has opened a new frontier in defining cell types within the intima. It will be important to use these methods to define the cell types comprising the atherosclerotic clone.
6. The extent of intimal cell masses across the normal human arterial tree of children is not known. There has been no thorough study of the arterial intima in the arterial tree of human children or other large mammals. Such masses may also occur later in life and lead to the development of new atherosclerotic lesions as suggested by DeBakey and Glaeser29.
7. Differences between models of atherosclerosis in mice versus the disease in humans may reflect the fact that mice lack intimal cells. Intimal cell masses, probably clonal in origin, may provide a soil for lesions that are distinct from those in the mouse model systems.
Grant information
Mark W. Majesky: National Institutes of Health grants HL123650; HL121877; HL133273; the Loie Power Robinson Stem Cell & Regenerative Medicine Fund, and the Seattle Children's Research Institute. Renu Virmani: Work is supported by the CVPath Institut.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Faculty Opinions recommendedReferences
- 1.
Benditt EP:
Evidence for a monoclonal origin of human atherosclerotic plaques and some implications.
Circulation.
1974; 50(4): 650–2. PubMed Abstract
| Publisher Full Text
- 2.
Chung IM, Schwartz SM, Murry CE:
Clonal architecture of normal and atherosclerotic aorta: implications for atherogenesis and vascular development.
Am J Pathol.
1998; 152(4): 913–23. PubMed Abstract
| Free Full Text
- 3.
Gomez D, Owens GK:
Reconciling Smooth Muscle Cell Oligoclonality and Proliferative Capacity in Experimental Atherosclerosis.
Circ Res.
2016; 119(12): 1262–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 4.
Ikari Y, McManus BM, Kenyon J, et al.:
Neonatal intima formation in the human coronary artery.
Arterioscler Thromb Vasc Biol.
1999; 19(9): 2036–40. PubMed Abstract
| Publisher Full Text
- 5.
Murry CE, Gipaya CT, Bartosek T, et al.:
Monoclonality of smooth muscle cells in human atherosclerosis.
Am J Pathol.
1997; 151(3): 697–705. PubMed Abstract
| Free Full Text
- 6.
Pearson TA, Dillman JM, Heptinstall RH:
Clonal mapping of the human aorta. Relationship of monoclonal characteristics, lesion thickness, and age in normal intima and atherosclerotic lesions.
Am J Pathol.
1987; 126(1): 33–9. PubMed Abstract
| Free Full Text
- 7.
Thomas WA, Lee KT, Kim DN:
Cell population kinetics in atherogenesis. Cell births and losses in intimal cell mass-derived lesions in the abdominal aorta of swine.
Ann N Y Acad Sci.
1985; 454: 305–15. PubMed Abstract
| Publisher Full Text
- 8.
Bentzon JF, Majesky MW:
Lineage tracking of origin and fate of smooth muscle cells in atherosclerosis.
Cardiovasc Res.
2018; 114(4): 492–500. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 9.
DiRenzo D, Owens GK, Leeper NJ:
"Attack of the Clones": Commonalities Between Cancer and Atherosclerosis.
Circ Res.
2017; 120(4): 624–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 10.
Benditt EP, Benditt JM:
Evidence for a monoclonal origin of human atherosclerotic plaques.
Proc Natl Acad Sci U S A.
1973; 70(6): 1753–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 11.
Moss NS, Benditt EP:
Human atherosclerotic plaque cells and leiomyoma cells. Comparison of in vitro growth characteristics.
Am J Pathol.
1975; 78(2): 175–90. PubMed Abstract
| Free Full Text
- 12.
Commandeur AE, Styer AK, Teixeira JM:
Epidemiological and genetic clues for molecular mechanisms involved in uterine leiomyoma development and growth.
Hum Reprod Update.
2015; 21(5): 593–615. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 13.
Dal Cin P, Moerman P, Deprest J, et al.:
A new cytogenetic subgroup in uterine leiomyoma is characterized by a deletion of the long arm of chromosome 3.
Genes Chromosomes Cancer.
1995; 13(3): 219–20. PubMed Abstract
| Publisher Full Text
- 14.
Fejzo MS, Yoon SJ, Montgomery KT, et al.:
Identification of a YAC spanning the translocation breakpoints in uterine leiomyomata, pulmonary chondroid hamartoma, and lipoma: physical mapping of the 12q14-q15 breakpoint region in uterine leiomyomata.
Genomics.
1995; 26(2): 265–71. PubMed Abstract
| Publisher Full Text
- 15.
Hashimoto K, Azuma C, Kamiura S, et al.:
Clonal determination of uterine leiomyomas by analyzing differential inactivation of the X-chromosome-linked phosphoglycerokinase gene.
Gynecol Obstet Invest.
1995; 40(3): 204–8. PubMed Abstract
| Publisher Full Text
- 16.
Hayashi S, Miharu N, Okamoto E, et al.:
Detection of chromosomal abnormalities of chromosome 12 in uterine leiomyoma using fluorescence in situ hybridization.
Jpn J Hum Genet.
1996; 41(1): 193–202. PubMed Abstract
| Publisher Full Text
- 17.
Hug K, Doney MK, Tyler MJ, et al.:
Physical mapping of the uterine leiomyoma t(12;14)(q13-15;q24.1) breakpoint on chromosome 14 between SPTB and D14S77.
Genes Chromosomes Cancer.
1994; 11(4): 263–6. PubMed Abstract
| Publisher Full Text
- 18.
Nilbert M, Heim S, Mandahl N, et al.:
Trisomy 12 in uterine leiomyomas. A new cytogenetic subgroup.
Cancer Genet Cytogenet.
1990; 45(1): 63–6. PubMed Abstract
| Publisher Full Text
- 19.
Vanni R, Lecca U, Faa G:
Uterine leiomyoma cytogenetics. II. Report of forty cases.
Cancer Genet Cytogenet.
1991; 53(2): 247–56. PubMed Abstract
| Publisher Full Text
- 20.
Williams AJ, Powell WL, Collins T, et al.:
HMGI(Y) expression in human uterine leiomyomata. Involvement of another high-mobility group architectural factor in a benign neoplasm.
Am J Pathol.
1997; 150(3): 911–8. PubMed Abstract
| Free Full Text
- 21.
Majesky MW, Reidy MA, Benditt EP, et al.:
Focal smooth muscle proliferation in the aortic intima produced by an initiation-promotion sequence.
Proc Natl Acad Sci U S A.
1985; 82(10): 3450–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 22.
Schwartz SM, Galis ZS, Rosenfeld ME, et al.:
Plaque rupture in humans and mice.
Arterioscler Thromb Vasc Biol.
2007; 27(4): 705–13. PubMed Abstract
| Publisher Full Text
- 23.
Yang X, Peterson L, Thieringer R, et al.:
Identification and validation of genes affecting aortic lesions in mice.
J Clin Invest.
2010; 120(7): 2414–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 24.
Stary HC:
Macrophage foam cells in the coronary artery intima of human infants.
Ann N Y Acad Sci.
1985; 454: 5–8. PubMed Abstract
| Publisher Full Text
- 25.
Yurovsky VV, Sutton PA, Schulze DH, et al.:
Expansion of selected V delta 1+ gamma delta T cells in systemic sclerosis patients.
J Immunol.
1994; 153(2): 881–91. PubMed Abstract
- 26.
Natural history of aortic and coronary atherosclerotic lesions in youth. Findings from the PDAY Study. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group.
Arterioscler Thromb.
1993; 13(9): 1291–8. PubMed Abstract
| Publisher Full Text
- 27.
Stary HC, Chandler AB, Dinsmore RE, et al.:
A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association.
Arterioscler Thromb Vasc Biol.
1995; 15(9): 1512–31. PubMed Abstract
| Publisher Full Text
- 28.
Stary HC, Chandler AB, Glagov S, et al.:
A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association.
Circulation.
1994; 89(5): 2462–78. PubMed Abstract
| Publisher Full Text
- 29.
DeBakey ME, Glaeser DH:
Patterns of atherosclerosis: effect of risk factors on recurrence and survival-analysis of 11,890 cases with more than 25-year follow-up.
Am J Cardiol.
2000; 85(9): 1045–53. PubMed Abstract
| Publisher Full Text
- 30.
Virchow R:
Cellular Pathology as Based Upon Physiological and Pathological Histology. Birmingham, Alabama: Classics of Medicine Library.
1858; 2. Reference Source
- 31.
Ross R:
Mechanisms of atherosclerosis--a review.
Adv Nephrol Necker Hosp.
1990; 19: 79–86. PubMed Abstract
- 32.
Getz GS, Reardon CA:
Animal models of atherosclerosis.
Arterioscler Thromb Vasc Biol.
2012; 32(5): 1104–15. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 33.
Getz GS, Reardon CA:
Use of Mouse Models in Atherosclerosis Research.
Methods Mol Biol.
2015; 1339: 1–16. PubMed Abstract
| Publisher Full Text
- 34.
Kaplan JR, Chen H, Manuck SB:
The relationship between social status and atherosclerosis in male and female monkeys as revealed by meta-analysis.
Am J Primatol.
2009; 71(9): 732–41. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 35.
Lusis AJ, Weiss JN:
Cardiovascular networks: systems-based approaches to cardiovascular disease.
Circulation.
2010; 121(1): 157–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 36.
Lusis AJ:
Atherosclerosis.
Nature.
2000; 407(6801): 233–41. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 37.
Ross R:
Rous-Whipple Award Lecture. Atherosclerosis: a defense mechanism gone awry.
Am J Pathol.
1993; 143(4): 987–1002. PubMed Abstract
| Free Full Text
- 38.
Sheth SS, Deluna A, Allayee H, et al.:
Understanding atherosclerosis through mouse genetics.
Curr Opin Lipidol.
2002; 13(2): 181–9. PubMed Abstract
| Publisher Full Text
- 39.
Shih DM, Welch C, Lusis AJ:
New insights into atherosclerosis from studies with mouse models.
Mol Med Today.
1995; 1(8): 364–72. PubMed Abstract
| Publisher Full Text
- 40.
Wang SS, Schadt EE, Wang H, et al.:
Identification of pathways for atherosclerosis in mice: integration of quantitative trait locus analysis and global gene expression data.
Circ Res.
2007; 101(3): e11–30. PubMed Abstract
| Publisher Full Text
- 41.
Wu S, Lusis AJ, Drake TA:
A systems-based framework for understanding complex metabolic and cardiovascular disorders.
J Lipid Res.
2009; 50(Suppl): S358–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 42.
Schwartz SM:
The intima: A new soil.
Circ Res.
1999; 85(10): 877–9. PubMed Abstract
- 43.
Schwartz SM, deBlois D, O'Brien ER:
The intima. Soil for atherosclerosis and restenosis.
Circ Res.
1995; 77(3): 445–65. PubMed Abstract
| Publisher Full Text
- 44.
Glass CK, Witztum JL:
Atherosclerosis. the road ahead.
Cell.
2001; 104(4): 503–16. PubMed Abstract
| Publisher Full Text
- 45.
Jacobsen K, Lund MB, Shim J, et al.:
Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs.
JCI Insight.
2017; 2(19): pii: 95890. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 46.
Bennett MR, Sinha S, Owens GK:
Vascular Smooth Muscle Cells in Atherosclerosis.
Circ Res.
2016; 118(4): 692–702. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 47.
Bentzon JF, Sondergaard CS, Kassem M, et al.:
Smooth muscle cells healing atherosclerotic plaque disruptions are of local, not blood, origin in apolipoprotein E knockout mice.
Circulation.
2007; 116(18): 2053–61. PubMed Abstract
| Publisher Full Text
- 48.
Chappell J, Harman JL, Narasimhan VM, et al.:
Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models.
Circ Res.
2016; 119(12): 1313–23. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 49.
Mazurek R, Dave JM, Chandran RR, et al.:
Vascular Cells in Blood Vessel Wall Development and Disease.
Adv Pharmacol.
2017; 78: 323–50. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 50.
Misra A, Feng Z, Chandran RR, et al.:
Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells.
Nat Commun.
2018; 9(1): 2073. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 51.
Cybulsky MI, Won D, Haidari M:
Leukocyte recruitment to atherosclerotic lesions.
Can J Cardiol.
2004; 20(Suppl B): 24B–28B. PubMed Abstract
- 52.
Rong JX, Shapiro M, Trogan E, et al.:
Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading.
Proc Natl Acad Sci U S A.
2003; 100(23): 13531–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 53.
Shankman LS, Gomez D, Cherepanova OA, et al.:
KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis.
Nat Med.
2015; 21(6): 628–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 54.
Shankman LS, Gomez D, Cherepanova OA, et al.:
KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis.
Nat Med.
2015; 21(6): 628–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 55.
Vengrenyuk Y, Nishi H, Long X, et al.:
Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype.
Arterioscler Thromb Vasc Biol.
2015; 35(3): 535–46. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 56.
Vanlandewijck M, He L, Mäe MA, et al.:
A molecular atlas of cell types and zonation in the brain vasculature.
Nature.
2018; 554(7693): 475–80. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 57.
Bargehr J, Low L, Cheung C, et al.:
Embryological Origin of Human Smooth Muscle Cells Influences Their Ability to Support Endothelial Network Formation.
Stem Cells Transl Med.
2016; 5(7): 946–59. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 58.
Majesky MW:
Developmental biology in the vasculature--review series.
Arterioscler Thromb Vasc Biol.
2009; 29(5): 622. PubMed Abstract
| Publisher Full Text
- 59.
Majesky MW:
Developmental basis of vascular smooth muscle diversity.
Arterioscler Thromb Vasc Biol.
2007; 27(6): 1248–58. PubMed Abstract
| Publisher Full Text
- 60.
Sinha S, Iyer D, Granata A:
Embryonic origins of human vascular smooth muscle cells: implications for in vitro modeling and clinical application.
Cell Mol Life Sci.
2014; 71(12): 2271–88. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 61.
Sinha S, Santoro MM:
New models to study vascular mural cell embryonic origin: implications in vascular diseases.
Cardiovasc Res.
2018; 114(4): 481–91. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 62.
Feil S, Hofmann F, Feil R:
SM22alpha modulates vascular smooth muscle cell phenotype during atherogenesis.
Circ Res.
2004; 94(7): 863–5. PubMed Abstract
| Publisher Full Text
- 63.
Wamhoff BR, Hoofnagle MH, Burns A, et al.:
A G/C element mediates repression of the SM22alpha promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis.
Circ Res.
2004; 95(10): 981–8. PubMed Abstract
| Publisher Full Text
- 64.
Firulli AB, Han D, Kelly-Roloff L, et al.:
A comparative molecular analysis of four rat smooth muscle cell lines.
In Vitro Cell Dev Biol Anim.
1998; 34(3): 217–26. PubMed Abstract
| Publisher Full Text
- 65.
Akyürek LM, Yang ZY, Aoki K, et al.:
SM22alpha promoter targets gene expression to vascular smooth muscle cells in vitro and in vivo.
Mol Med.
2000; 6(11): 983–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 66.
Albinsson S, Skoura A, Yu J, et al.:
Smooth muscle miRNAs are critical for post-natal regulation of blood pressure and vascular function.
PLoS One.
2011; 6(4): e18869. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 67.
Bär H, Wende P, Watson L, et al.:
Smoothelin is an indicator of reversible phenotype modulation of smooth muscle cells in balloon-injured rat carotid arteries.
Basic Res Cardiol.
2002; 97(1): 9–16. PubMed Abstract
| Publisher Full Text
- 68.
Cai WJ, Kocsis E, Scholz D, et al.:
Presence of Cx37 and lack of desmin in smooth muscle cells are early markers for arteriogenesis.
Mol Cell Biochem.
2004; 262(1–2): 17–23. PubMed Abstract
| Publisher Full Text
- 69.
Camoretti-Mercado B, Liu HW, Halayko AJ, et al.:
Physiological control of smooth muscle-specific gene expression through regulated nuclear translocation of serum response factor.
J Biol Chem.
2000; 275(39): 30387–93. PubMed Abstract
| Publisher Full Text
- 70.
Chan-Ling T, Koina ME, McColm JR, et al.:
Role of CD44+ stem cells in mural cell formation in the human choroid: evidence of vascular instability due to limited pericyte ensheathment.
Invest Ophthalmol Vis Sci.
2011; 52(1): 399–410. PubMed Abstract
| Publisher Full Text
- 71.
Duband JL, Gimona M, Scatena M, et al.:
Calponin and SM 22 as differentiation markers of smooth muscle: spatiotemporal distribution during avian embryonic development.
Differentiation.
1993; 55(1): 1–11. PubMed Abstract
| Publisher Full Text
- 72.
Frid MG, Moiseeva EP, Stenmark KR:
Multiple phenotypically distinct smooth muscle cell populations exist in the adult and developing bovine pulmonary arterial media in vivo.
Circ Res.
1994; 75(4): 669–81. PubMed Abstract
| Publisher Full Text
- 73.
Frid MG, Shekhonin BV, Koteliansky VE, et al.:
Phenotypic changes of human smooth muscle cells during development: late expression of heavy caldesmon and calponin.
Dev Biol.
1992; 153(2): 185–93. PubMed Abstract
| Publisher Full Text
- 74.
Gabbiani G, Kocher O, Bloom WS, et al.:
Actin expression in smooth muscle cells of rat aortic intimal thickening, human atheromatous plaque, and cultured rat aortic media.
J Clin Invest.
1984; 73(1): 148–52. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 75.
Galmiche MC, Koteliansky VE, Brière J, et al.:
Stromal cells from human long-term marrow cultures are mesenchymal cells that differentiate following a vascular smooth muscle differentiation pathway.
Blood.
1993; 82(1): 66–76. PubMed Abstract
- 76.
Gröschel-Stewart U, Chamley JH, McConnell JD, et al.:
Comparison of the reaction of cultured smooth and cardiac muscle cells and fibroblasts to specific antibodies to myosin.
Histochemistry.
1975; 43(3): 215–24. PubMed Abstract
| Publisher Full Text
- 77.
Johansson B, Eriksson A, Ramaekers F, et al.:
Smoothelin in adult and developing human arteries and myocardium.
Histochem Cell Biol.
1999; 112(4): 291–9. PubMed Abstract
| Publisher Full Text
- 78.
Johansson B, Eriksson A, Ramaekers F, et al.:
Smoothelin and intermediate filament proteins in human aortocoronary saphenous vein by-pass grafts.
Histochem J.
1999; 31(11): 723–7. PubMed Abstract
| Publisher Full Text
- 79.
Kacem K, Seylaz J, Aubineau P:
Differential processes of vascular smooth muscle cell differentiation within elastic and muscular arteries of rats and rabbits: an immunofluorescence study of desmin and vimentin distribution.
Histochem J.
1996; 28(1): 53–61. PubMed Abstract
| Publisher Full Text
- 80.
Kaplan-Albuquerque N, Garat C, Van Putten V, et al.:
Regulation of SM22 alpha expression by arginine vasopressin and PDGF-BB in vascular smooth muscle cells.
Am J Physiol Heart Circ Physiol.
2003; 285(4): H1444–52. PubMed Abstract
| Publisher Full Text
- 81.
Ko YS, Coppen SR, Dupont E, et al.:
Regional differentiation of desmin, connexin43, and connexin45 expression patterns in rat aortic smooth muscle.
Arterioscler Thromb Vasc Biol.
2001; 21(3): 355–64. PubMed Abstract
| Publisher Full Text
- 82.
Ko YS, Yeh HI, Haw M, et al.:
Differential expression of connexin43 and desmin defines two subpopulations of medial smooth muscle cells in the human internal mammary artery.
Arterioscler Thromb Vasc Biol.
1999; 19(7): 1669–80. PubMed Abstract
| Publisher Full Text
- 83.
Kolodziejska KM, Noyan-Ashraf MH, Nagy A, et al.:
c-Myb-dependent smooth muscle cell differentiation.
Circ Res.
2008; 102(5): 554–61. PubMed Abstract
| Publisher Full Text
- 84.
Landerholm TE, Dong XR, Lu J, et al.:
A role for serum response factor in coronary smooth muscle differentiation from proepicardial cells.
Development.
1999; 126(10): 2053–62. PubMed Abstract
- 85.
Lees-Miller JP, Heeley DH, Smillie LB:
An abundant and novel protein of 22 kDa (SM22) is widely distributed in smooth muscles. Purification from bovine aorta.
Biochem J.
1987; 244(3): 705–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 86.
Li L, Miano JM, Mercer B, et al.:
Expression of the SM22alpha promoter in transgenic mice provides evidence for distinct transcriptional regulatory programs in vascular and visceral smooth muscle cells.
J Cell Biol.
1996; 132(5): 849–59. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 87.
Li ZL, Lilienbaum A, Butler-Browne G, et al.:
Human desmin-coding gene: complete nucleotide sequence, characterization and regulation of expression during myogenesis and development.
Gene.
1989; 78(2): 243–54. PubMed Abstract
| Publisher Full Text
- 88.
Mack CP, Somlyo AV, Hautmann M, et al.:
Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization.
J Biol Chem.
2001; 276(1): 341–7. PubMed Abstract
| Publisher Full Text
- 89.
Mericskay M, Li Z, Paulin D:
Transcriptional regulation of the desmin and SM22 genes in vascular smooth muscle cells.
Curr Top Pathol.
1999; 93: 7–17. PubMed Abstract
| Publisher Full Text
- 90.
Miano JM, Cserjesi P, Ligon KL, et al.:
Smooth muscle myosin heavy chain exclusively marks the smooth muscle lineage during mouse embryogenesis.
Circ Res.
1994; 75(5): 803–12. PubMed Abstract
| Publisher Full Text
- 91.
Miano JM, Olson EN:
Expression of the smooth muscle cell calponin gene marks the early cardiac and smooth muscle cell lineages during mouse embryogenesis.
J Biol Chem.
1996; 271(12): 7095–103. PubMed Abstract
| Publisher Full Text
- 92.
Moessler H, Mericskay M, Li Z, et al.:
The SM 22 promoter directs tissue-specific expression in arterial but not in venous or visceral smooth muscle cells in transgenic mice.
Development.
1996; 122(8): 2415–25. PubMed Abstract
- 93.
Rensen SS, Thijssen VL, De Vries CJ, et al.:
Expression of the smoothelin gene is mediated by alternative promoters.
Cardiovasc Res.
2002; 55(4): 850–63. PubMed Abstract
| Publisher Full Text
- 94.
Ruzicka DL, Schwartz RJ:
Sequential activation of alpha-actin genes during avian cardiogenesis: vascular smooth muscle alpha-actin gene transcripts mark the onset of cardiomyocyte differentiation.
J Cell Biol.
1988; 107(6 Pt 2): 2575–86. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 95.
Samaha FF, Ip HS, Morrisey EE, et al.:
Developmental pattern of expression and genomic organization of the calponin-h1 gene. A contractile smooth muscle cell marker.
J Biol Chem.
1996; 271(1): 395–403. PubMed Abstract
| Publisher Full Text
- 96.
Sjuve R, Arner A, Li Z, et al.:
Mechanical alterations in smooth muscle from mice lacking desmin.
J Muscle Res Cell Motil.
1998; 19(4): 415–29. PubMed Abstract
| Publisher Full Text
- 97.
Skalli O, Gabbiani F, Gabbiani G:
Action of general and alpha-smooth muscle-specific actin antibody microinjection on stress fibers of cultured smooth muscle cells.
Exp Cell Res.
1990; 187(1): 119–25. PubMed Abstract
| Publisher Full Text
- 98.
Skalli O, Ropraz P, Trzeciak A, et al.:
A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation.
J Cell Biol.
1986; 103(6 Pt 2): 2787–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 99.
Sobue K, Hayashi K, Nishida W:
Expressional regulation of smooth muscle cell-specific genes in association with phenotypic modulation.
Mol Cell Biochem.
1999; 190(1–2): 105–18. PubMed Abstract
| Publisher Full Text
- 100.
Solway J, Seltzer J, Samaha FF, et al.:
Structure and expression of a smooth muscle cell-specific gene, SM22 alpha.
J Biol Chem.
1995; 270(22): 13460–9. PubMed Abstract
| Publisher Full Text
- 101.
Takahashi K, Hiwada K, Kokubu T:
Vascular smooth muscle calponin. A novel troponin T-like protein.
Hypertension.
1988; 11(6 Pt 2): 620–6. PubMed Abstract
| Publisher Full Text
- 102.
van der Loop FT, Gabbiani G, Kohnen G, et al.:
Differentiation of smooth muscle cells in human blood vessels as defined by smoothelin, a novel marker for the contractile phenotype.
Arterioscler Thromb Vasc Biol.
1997; 17(4): 665–71. PubMed Abstract
| Publisher Full Text
- 103.
van Eys GJ, Völler MC, Timmer ED, et al.:
Smoothelin expression characteristics: development of a smooth muscle cell in vitro system and identification of a vascular variant.
Cell Struct Funct.
1997; 22(1): 65–72. PubMed Abstract
| Publisher Full Text
- 104.
Wehrens XH, Mies B, Gimona M, et al.:
Localization of smoothelin in avian smooth muscle and identification of a vascular-specific isoform.
FEBS Lett.
1997; 405(3): 315–20. PubMed Abstract
| Publisher Full Text
- 105.
Wirth A, Benyó Z, Lukasova M, et al.:
G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension.
Nat Med.
2008; 14(1): 64–8. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 106.
Wu SP, Dong XR, Regan JN, et al.:
Tbx18 regulates development of the epicardium and coronary vessels.
Dev Biol.
2013; 383(2): 307–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 107.
Xu R, Ho YS, Ritchie RP, et al.:
Human SM22 alpha BAC encompasses regulatory sequences for expression in vascular and visceral smooth muscles at fetal and adult stages.
Am J Physiol Heart Circ Physiol.
2003; 284(4): H1398–407. PubMed Abstract
| Publisher Full Text
- 108.
Yablonka-Reuveni Z, Christ B, Benson JM:
Transitions in cell organization and in expression of contractile and extracellular matrix proteins during development of chicken aortic smooth muscle: evidence for a complex spatial and temporal differentiation program.
Anat Embryol (Berl).
1998; 197(6): 421–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 109.
Zhang JC, Kim S, Helmke BP, et al.:
Analysis of SM22alpha-deficient mice reveals unanticipated insights into smooth muscle cell differentiation and function.
Mol Cell Biol.
2001; 21(4): 1336–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 110.
Zohlnhöfer D, Klein CA, Richter T, et al.:
Gene expression profiling of human stent-induced neointima by cDNA array analysis of microscopic specimens retrieved by helix cutter atherectomy: Detection of FK506-binding protein 12 upregulation.
Circulation.
2001; 103(10): 1396–402. PubMed Abstract
| Publisher Full Text
- 111.
Andrews A, Balciunaite E, Leong FL, et al.:
Platelet-derived growth factor plays a key role in proliferative vitreoretinopathy.
Invest Ophthalmol Vis Sci.
1999; 40(11): 2683–9. PubMed Abstract
- 112.
Armulik A, Genové G, Betsholtz C:
Pericytes: developmental, physiological, and pathological perspectives, problems, and promises.
Dev Cell.
2011; 21(2): 193–215. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 113.
Benjamin LE, Hemo I, Keshet E:
A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF.
Development.
1998; 125(9): 1591–8. PubMed Abstract
| Faculty Opinions Recommendation
- 114.
Berger M, Bergers G, Arnold B, et al.:
Regulator of G-protein signaling-5 induction in pericytes coincides with active vessel remodeling during neovascularization.
Blood.
2005; 105(3): 1094–101. PubMed Abstract
| Publisher Full Text
- 115.
Bondjers C, He L, Takemoto M, et al.:
Microarray analysis of blood microvessels from PDGF-B and PDGF-Rbeta mutant mice identifies novel markers for brain pericytes.
FASEB J.
2006; 20(10): 1703–5. PubMed Abstract
| Publisher Full Text
- 116.
Bondjers C, Kalén M, Hellström M, et al.:
Transcription profiling of platelet-derived growth factor-B-deficient mouse embryos identifies RGS5 as a novel marker for pericytes and vascular smooth muscle cells.
Am J Pathol.
2003; 162(3): 721–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 117.
Chen YT, Chang FC, Wu CF, et al.:
Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis.
Kidney Int.
2011; 80(11): 1170–81. PubMed Abstract
| Publisher Full Text
- 118.
Cho H, Kozasa T, Bondjers C, et al.:
Pericyte-specific expression of Rgs5: implications for PDGF and EDG receptor signaling during vascular maturation.
FASEB J.
2003; 17(3): 440–2. PubMed Abstract
| Publisher Full Text
- 119.
Cho H, Park C, Hwang IY, et al.:
Rgs5 targeting leads to chronic low blood pressure and a lean body habitus.
Mol Cell Biol.
2008; 28(8): 2590–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 120.
Crisan M, Yap S, Casteilla L, et al.:
A perivascular origin for mesenchymal stem cells in multiple human organs.
Cell Stem Cell.
2008; 3(3): 301–13. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 121.
Dhar K, Dhar G, Majumder M, et al.:
Tumor cell-derived PDGF-B potentiates mouse mesenchymal stem cells-pericytes transition and recruitment through an interaction with NRP-1.
Mol Cancer.
2010; 9: 209. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 122.
Hellström M, Gerhardt H, Kalén M, et al.:
Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis.
J Cell Biol.
2001; 153(3): 543–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 123.
Herman IM, D'Amore PA:
Microvascular pericytes contain muscle and nonmuscle actins.
J Cell Biol.
1985; 101(1): 43–52. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 124.
Howson KM, Aplin AC, Gelati M, et al.:
The postnatal rat aorta contains pericyte progenitor cells that form spheroidal colonies in suspension culture.
Am J Physiol Cell Physiol.
2005; 289(6): C1396–407. PubMed Abstract
| Publisher Full Text
- 125.
Mitchell TS, Bradley J, Robinson GS, et al.:
RGS5 expression is a quantitative measure of pericyte coverage of blood vessels.
Angiogenesis.
2008; 11(2): 141–51. PubMed Abstract
| Publisher Full Text
- 126.
Ozerdem U, Grako KA, Dahlin-Huppe K, et al.:
NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis.
Dev Dyn.
2001; 222(2): 218–27. PubMed Abstract
| Publisher Full Text
- 127.
Schrimpf C, Duffield JS:
Mechanisms of fibrosis: the role of the pericyte.
Curr Opin Nephrol Hypertens.
2011; 20(3): 297–305. PubMed Abstract
| Publisher Full Text
- 128.
Moore-Morris T, Tallquist MD, Evans SM:
Sorting out where fibroblasts come from.
Circ Res.
2014; 115(7): 602–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 129.
Snider P, Hinton RB, Moreno-Rodriguez RA, et al.:
Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart.
Circ Res.
2008; 102(7): 752–60. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 130.
Denton CP, Zheng B, Shiwen X, et al.:
Activation of a fibroblast-specific enhancer of the proalpha2(I) collagen gene in tight-skin mice.
Arthritis Rheum.
2001; 44(3): 712–22. PubMed Abstract
| Publisher Full Text
- 131.
Gebhard S, Hattori T, Bauer E, et al.:
Specific expression of Cre recombinase in hypertrophic cartilage under the control of a BAC-Col10a1 promoter.
Matrix Biol.
2008; 27(8): 693–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 132.
Iwano M, Fischer A, Okada H, et al.:
Conditional abatement of tissue fibrosis using nucleoside analogs to selectively corrupt DNA replication in transgenic fibroblasts.
Mol Ther.
2001; 3(2): 149–59. PubMed Abstract
| Publisher Full Text
- 133.
Niederreither K, D'Souza RN, de Crombrugghe B:
Minimal DNA sequences that control the cell lineage-specific expression of the pro alpha 2(I) collagen promoter in transgenic mice.
J Cell Biol.
1992; 119(5): 1361–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 134.
Soundararajan M, Kannan S:
Fibroblasts and mesenchymal stem cells: Two sides of the same coin?
J Cell Physiol.
2018; 233(12): 9099–109. PubMed Abstract
| Publisher Full Text
- 135.
Wu J, Montaniel KR, Saleh MA, et al.:
Origin of Matrix-Producing Cells That Contribute to Aortic Fibrosis in Hypertension.
Hypertension.
2016; 67(2): 461–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 136.
Hu Y, Zhang Z, Torsney E, et al.:
Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice.
J Clin Invest.
2004; 113(9): 1258–65. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 137.
Passman JN, Dong XR, Wu SP, et al.:
A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells.
Proc Natl Acad Sci U S A.
2008; 105(27): 9349–54. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 138.
Harhun MI, Pucovský V, Povstyan OV, et al.:
Interstitial cells in the vasculature.
J Cell Mol Med.
2005; 9(2): 232–43. PubMed Abstract
| Publisher Full Text
- 139.
Hibbert B, Chen YX, O'Brien ER:
c-kit-immunopositive vascular progenitor cells populate human coronary in-stent restenosis but not primary atherosclerotic lesions.
Am J Physiol Heart Circ Physiol.
2004; 287(2): H518–24. PubMed Abstract
| Publisher Full Text
- 140.
Kramann R, Schneider RK, DiRocco DP, et al.:
Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis.
Cell Stem Cell.
2015; 16(1): 51–66. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 141.
Zengin E, Chalajour F, Gehling UM, et al.:
Vascular wall resident progenitor cells: a source for postnatal vasculogenesis.
Development.
2006; 133(8): 1543–51. PubMed Abstract
| Publisher Full Text
- 142.
Fleming JN, Nash RA, Mahoney WM Jr, et al.:
Is scleroderma a vasculopathy?
Curr Rheumatol Rep.
2009; 11(2): 103–10. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 143.
Muro AF, Moretti FA, Moore BB, et al.:
An essential role for fibronectin extra type III domain A in pulmonary fibrosis.
Am J Respir Crit Care Med.
2008; 177(6): 638–45. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 144.
Rönty MJ, Leivonen SK, Hinz B, et al.:
Isoform-specific regulation of the actin-organizing protein palladin during TGF-beta1-induced myofibroblast differentiation.
J Invest Dermatol.
2006; 126(11): 2387–96. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 145.
Schürch W, Seemayer TA, Lagacé R, et al.:
The intermediate filament cytoskeleton of myofibroblasts: an immunofluorescence and ultrastructural study.
Virchows Arch A Pathol Anat Histopathol.
1984; 403(4): 323–36. PubMed Abstract
| Publisher Full Text
- 146.
Skalli O, Schürch W, Seemayer T, et al.:
Myofibroblasts from diverse pathologic settings are heterogeneous in their content of actin isoforms and intermediate filament proteins.
Lab Invest.
1989; 60(2): 275–85. PubMed Abstract
- 147.
Kovanen PT, Tabas I:
In search of a starting point.
Curr Opin Lipidol.
2001; 12(5): 475–6. PubMed Abstract
- 148.
Feil S, Fehrenbacher B, Lukowski R, et al.:
Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis.
Circ Res.
2014; 115(7): 662–7. PubMed Abstract
| Publisher Full Text
- 149.
Wang J, Uryga AK, Reinhold J, et al.:
Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability.
Circulation.
2015; 132(20): 1909–19. PubMed Abstract
| Publisher Full Text
- 150.
Doney MK, Gerken SC, Lynch R, et al.:
Precise mapping of t(12;14) leiomyoma breakpoint on chromosome 14 between D14S298 and D14S540.
Cancer Lett.
1995; 96(2): 245–52. PubMed Abstract
| Publisher Full Text
- 151.
Hayashi S, Miharu N, Okamoto E, et al.:
Detection of chromosomal abnormalities in uterine leiomyoma using conventional cytogenetic method and interphase fluorescence in situ hybridization.
Cancer Genet Cytogenet.
1996; 89(2): 98–104. PubMed Abstract
| Publisher Full Text
- 152.
Karaiskos C, Pandis N, Bardi G, et al.:
Cytogenetic findings in uterine epithelioid leiomyomas.
Cancer Genet Cytogenet.
1995; 80(2): 103–6. PubMed Abstract
| Publisher Full Text
- 153.
Matsuo H, Maruo T, Samoto T:
Increased expression of Bcl-2 protein in human uterine leiomyoma and its up-regulation by progesterone.
J Clin Endocrinol Metab.
1997; 82(1): 293–9. PubMed Abstract
| Publisher Full Text
- 154.
Nilbert M, Rydholm A, Mitelman F, et al.:
Characterization of the 12q13-15 amplicon in soft tissue tumors.
Cancer Genet Cytogenet.
1995; 83(1): 32–6. PubMed Abstract
| Publisher Full Text
- 155.
Sourla A, Polychronakos C, Zeng WR, et al.:
Plasminogen activator inhibitor 1 messenger RNA expression and molecular evidence for del(7)(q22) in uterine leiomyomas.
Cancer Res.
1996; 56(13): 3123–8. PubMed Abstract
- 156.
Valenti MT, Azzarello G, Vinante O, et al.:
Differentiation, proliferation and apoptosis levels in human leiomyoma and leiomyosarcoma.
J Cancer Res Clin Oncol.
1998; 124(2): 93–105. PubMed Abstract
| Publisher Full Text
- 157.
Van de Ven WJ, Schoenmakers EF, Wanschura S, et al.:
Molecular characterization of MAR, a multiple aberration region on human chromosome segment 12q13-q15 implicated in various solid tumors.
Genes Chromosomes Cancer.
1995; 12(4): 296–303. PubMed Abstract
| Publisher Full Text
- 158.
Wanschura S, Hennig Y, Deichert U, et al.:
Molecular-cytogenetic refinement of the 12q14-->q15 breakpoint region affected in uterine leiomyomas.
Cytogenet Cell Genet.
1995; 71(2): 131–5. PubMed Abstract
| Publisher Full Text
- 159.
Krug EL, Rezaee M, Isokawa K, et al.:
Transformation of cardiac endothelium into cushion mesenchyme is dependent on ES/130: Temporal, spatial, and functional studies in the early chick embryo.
Cell Mol Biol Res.
1995; 41(4): 263–77. PubMed Abstract
- 160.
Sugi Y, Markwald RR:
Formation and early morphogenesis of endocardial endothelial precursor cells and the role of endoderm.
Dev Biol.
1996; 175(1): 66–83. PubMed Abstract
| Publisher Full Text
- 161.
Chappell D, Jacob M, Paul O, et al.:
The glycocalyx of the human umbilical vein endothelial cell: an impressive structure ex vivo but not in culture.
Circ Res.
2009; 104(11): 1313–7. PubMed Abstract
| Publisher Full Text
- 162.
Plein A, Fantin A, Denti L, et al.:
Erythro-myeloid progenitors contribute endothelial cells to blood vessels.
Nature.
2018; 562(7726): 223–8. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 163.
Yang Q, Mas A, Diamond MP, et al.:
The Mechanism and Function of Epigenetics in Uterine Leiomyoma Development.
Reprod Sci.
2016; 23(2): 163–75. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 164.
Majesky MW, Dong XR, Regan JN, et al.:
Vascular smooth muscle progenitor cells: building and repairing blood vessels.
Circ Res.
2011; 108(3): 365–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 165.
Lu J, Landerholm TE, Wei JS, et al.:
Coronary smooth muscle differentiation from proepicardial cells requires rhoA-mediated actin reorganization and p160 rho-kinase activity.
Dev Biol.
2001; 240(2): 404–18. PubMed Abstract
| Publisher Full Text
- 166.
Majesky MW:
Vascular smooth muscle diversity: insights from developmental biology.
Curr Atheroscler Rep.
2003; 5(3): 208–13. PubMed Abstract
| Publisher Full Text
- 167.
Majesky MW, Schwartz SM:
Smooth muscle diversity in arterial wound repair.
Toxicol Pathol.
1990; 18(4 Pt 1): 554–9. PubMed Abstract
- 168.
Schwartz SM, Heimark RL, Majesky MW:
Developmental mechanisms underlying pathology of arteries.
Physiol Rev.
1990; 70(4): 1177–209. PubMed Abstract
| Publisher Full Text
- 169.
Topouzis S, Majesky MW:
Smooth muscle lineage diversity in the chick embryo. Two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-beta.
Dev Biol.
1996; 178(2): 430–45. PubMed Abstract
| Publisher Full Text
- 170.
Mikawa T, Fischman DA:
Retroviral analysis of cardiac morphogenesis: Discontinuous formation of coronary vessels.
Proc Natl Acad Sci U S A.
1992; 89(20): 9504–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 171.
Flouris GA, Arvanitis DA, Parissis JT, et al.:
Loss of heterozygosity in DNA mismatch repair genes in human atherosclerotic plaques.
Mol Cell Biol Res Commun.
2000; 4(1): 62–5. PubMed Abstract
| Publisher Full Text
- 172.
Kiaris H, Hatzistamou J, Spandidos DA:
Instability at the H-ras minisatellite in human atherosclerotic plaques.
Atherosclerosis.
1996; 125(1): 47–51. PubMed Abstract
| Publisher Full Text
- 173.
Pearson TA, Dillman JM, Solez K, et al.:
Clonal characteristics in layers of human atherosclerotic plaques. A study of the selection hypothesis of monoclonality.
Am J Pathol.
1978; 93(1): 93–102. PubMed Abstract
| Free Full Text
- 174.
Pearson TA, Dillman JM, Solez K, et al.:
Clonal characteristics of cutaneous scars and implications for atherogenesis.
Am J Pathol.
1981; 102(1): 49–54. PubMed Abstract
| Free Full Text
- 175.
Pearson TA, Dillman J, Solez K, et al.:
Monoclonal characteristics of organising arterial thrombi: Significance in the origin and growth of human atherosclerotic plaques.
Lancet.
1979; 1(8106): 7–11. PubMed Abstract
| Publisher Full Text
- 176.
Schwartz SM, Murry CE:
Proliferation and the monoclonal origins of atherosclerotic lesions.
Annu Rev Med.
1998; 49: 437–60. PubMed Abstract
| Publisher Full Text
- 177.
Wagner DE, Weinreb C, Collins ZM, et al.:
Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo.
Science.
2018; 360(6392): 981–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 178.
Chung IM, et al.:
Monoclonality in atherosclerosis may arise by expansion of a pre-existing clone of smooth muscle cells.
Circulation.
1996; 94(Suppl 1): 1–238.
- 179.
Salipante SJ, Thompson JM, Horwitz MS:
Phylogenetic fate mapping: theoretical and experimental studies applied to the development of mouse fibroblasts.
Genetics.
2008; 178(2): 967–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 180.
Horwitz PA, Tsai EJ, Putt ME, et al.:
Detection of cardiac allograft rejection and response to immunosuppressive therapy with peripheral blood gene expression.
Circulation.
2004; 110(25): 3815–21. PubMed Abstract
| Publisher Full Text
- 181.
Risques RA, Kennedy SR:
Aging and the rise of somatic cancer-associated mutations in normal tissues.
PLoS Genet.
2018; 14(1): e1007108. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 182.
Bedrosian TA, Quayle C, Novaresi N, et al.:
Early life experience drives structural variation of neural genomes in mice.
Science.
2018; 359(6382): 1395–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 183.
Insel TR:
Brain somatic mutations: the dark matter of psychiatric genetics?
Mol Psychiatry.
2014; 19(2): 156–8. PubMed Abstract
| Publisher Full Text
- 184.
Paquola ACM, Erwin JA, Gage FH:
Insights into the role of somatic mosaicism in the brain.
Curr Opin Syst Biol.
2017; 1: 90–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 185.
Andreassi MG, Botto N, Colombo MG, et al.:
Genetic instability and atherosclerosis: can somatic mutations account for the development of cardiovascular diseases?
Environ Mol Mutagen.
2000; 35(4): 265–9. PubMed Abstract
- 186.
Ballinger SW, Patterson C, Knight-Lozano CA, et al.:
Mitochondrial integrity and function in atherogenesis.
Circulation.
2002; 106(5): 544–9. PubMed Abstract
| Publisher Full Text
- 187.
Bennett MR:
Reactive oxygen species and death: oxidative DNA damage in atherosclerosis.
Circ Res.
2001; 88(7): 648–50. PubMed Abstract
| Publisher Full Text
- 188.
Binková B, Smerhovský Z, Strejc P, et al.:
DNA-adducts and atherosclerosis: a study of accidental and sudden death males in the Czech Republic.
Mutat Res.
2002; 501(1–2): 115–28. PubMed Abstract
| Publisher Full Text
- 189.
Borgia MC, Mandolini C, Barresi C, et al.:
Further evidence against the implication of active cytomegalovirus infection in vascular atherosclerotic diseases.
Atherosclerosis.
2001; 157(2): 457–62. PubMed Abstract
| Publisher Full Text
- 190.
Botto N, Masetti S, Petrozzi L, et al.:
Elevated levels of oxidative DNA damage in patients with coronary artery disease.
Coron Artery Dis.
2002; 13(5): 269–74. PubMed Abstract
| Publisher Full Text
- 191.
Botto N, Rizza A, Colombo MG, et al.:
Evidence for DNA damage in patients with coronary artery disease.
Mutat Res.
2001; 493(1–2): 23–30. PubMed Abstract
| Publisher Full Text
- 192.
Chiu B:
Multiple infections in carotid atherosclerotic plaques.
Am Heart J.
1999; 138(5 Pt 2): S534–6. PubMed Abstract
| Publisher Full Text
- 193.
Clark KJ, Cary NR, Grace AA, et al.:
Microsatellite mutation of type II transforming growth factor-beta receptor is rare in atherosclerotic plaques.
Arterioscler Thromb Vasc Biol.
2001; 21(4): 555–9. PubMed Abstract
| Publisher Full Text
- 194.
Corral-Debrinski M, Shoffner JM, Lott MT, et al.:
Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease.
Mutat Res.
1992; 275(3–6): 169–80. PubMed Abstract
| Publisher Full Text
- 195.
Fuster JJ, MacLauchlan S, Zuriaga MA, et al.:
Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice.
Science.
2017; 355(6327): 842–847. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 196.
Grati FR, Ghilardi G, Sirchia SM, et al.:
Loss of heterozygosity of the NOS3 dinucleotide repeat marker in atherosclerotic plaques of human carotid arteries.
Atherosclerosis.
2001; 159(2): 261–7. PubMed Abstract
| Publisher Full Text
- 197.
Hatzistamou J, Kiaris H, Ergazaki M, et al.:
Loss of heterozygosity and microsatellite instability in human atherosclerotic plaques.
Biochem Biophys Res Commun.
1996; 225(1): 186–90. PubMed Abstract
| Publisher Full Text
- 198.
Hemnani T, Parihar MS:
Reactive oxygen species and oxidative DNA damage.
Indian J Physiol Pharmacol.
1998; 42(4): 440–52. PubMed Abstract
- 199.
Hendrix MG, Daemen M, Bruggeman CA:
Cytomegalovirus nucleic acid distribution within the human vascular tree.
Am J Pathol.
1991; 138(3): 563–7. PubMed Abstract
| Free Full Text
- 200.
Hendrix MG, Dormans PH, Kitslaar P, et al.:
The presence of cytomegalovirus nucleic acids in arterial walls of atherosclerotic and nonatherosclerotic patients.
Am J Pathol.
1989; 134(5): 1151–7. PubMed Abstract
| Free Full Text
- 201.
Horváth R, Cerný J, Benedík J Jr, et al.:
The possible role of human cytomegalovirus (HCMV) in the origin of atherosclerosis.
J Clin Virol.
2000; 16(1): 17–24. PubMed Abstract
| Publisher Full Text
- 202.
Izzotti A, Cartiglia C, Lewtas J, et al.:
Increased DNA alterations in atherosclerotic lesions of individuals lacking the GSTM1 genotype.
FASEB J.
2001; 15(3): 752–7. PubMed Abstract
| Publisher Full Text
- 203.
Kreja L, Finking G:
Development of an in vitro model to study oxidative DNA damage in human coronary smooth muscle cells.
ALTEX.
2002; 19(3): 123–9. PubMed Abstract
- 204.
Liao S, Curci JA, Kelley BJ, et al.:
Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery.
J Surg Res.
2000; 92(1): 85–95. PubMed Abstract
| Publisher Full Text
- 205.
Martinet W, Knaapen MW, De Meyer GR, et al.:
Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering.
Circ Res.
2001; 88(7): 733–9. PubMed Abstract
| Publisher Full Text
- 206.
Martinet W, Knaapen MW, De Meyer GR, et al.:
Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques.
Circulation.
2002; 106(8): 927–32. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 207.
Melnick JL, Adam E, DeBakey ME:
Cytomegalovirus and atherosclerosis.
Arch Immunol Ther Exp (Warsz).
1996; 44(5–6): 297–302. PubMed Abstract
- 208.
Miniati P, Sourvinos G, Michalodimitrakis M, et al.:
Loss of heterozygosity on chromosomes 1, 2, 8, 9 and 17 in cerebral atherosclerotic plaques.
Int J Biol Markers.
2001; 16(3): 167–71. PubMed Abstract
| Publisher Full Text
- 209.
Olinski R, Gackowski D, Foksinski M, et al.:
Oxidative DNA damage: assessment of the role in carcinogenesis, atherosclerosis, and acquired immunodeficiency syndrome.
Free Radic Biol Med.
2002; 33(2): 192–200. PubMed Abstract
| Publisher Full Text
- 210.
Parkes JL, Cardell RR, Hubbard FC Jr, et al.:
Cultured human atherosclerotic plaque smooth muscle cells retain transforming potential and display enhanced expression of the myc protooncogene.
Am J Pathol.
1991; 138(3): 765–75. PubMed Abstract
| Free Full Text
- 211.
Saetta A, Fanourakis G, Agapitos E, et al.:
Atherosclerosis of the carotid artery: absence of evidence for CMV involvement in atheroma formation.
Cardiovasc Pathol.
2000; 9(3): 181–3. PubMed Abstract
| Publisher Full Text
- 212.
Shi Y, Tokunaga O:
Herpesvirus (HSV-1, EBV and CMV) infections in atherosclerotic compared with non-atherosclerotic aortic tissue.
Pathol Int.
2002; 52(1): 31–9. PubMed Abstract
| Publisher Full Text
- 213.
Spandidos DA, Ergazaki M, Arvanitis D, et al.:
Microsatellite instability in human atherosclerotic plaques.
Biochem Biophys Res Commun.
1996; 220(1): 137–40. PubMed Abstract
| Publisher Full Text
- 214.
Weakley SM, Jiang J, Kougias P, et al.:
Role of somatic mutations in vascular disease formation.
Expert Rev Mol Diagn.
2010; 10(2): 173–85. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 215.
Yamashiroya HM, Ghosh L, Yang R, et al.:
Herpesviridae in the coronary arteries and aorta of young trauma victims.
Am J Pathol.
1988; 130(1): 71–9. PubMed Abstract
| Free Full Text
- 216.
Zhou YF, Shou M, Guetta E, et al.:
Cytomegalovirus infection of rats increases the neointimal response to vascular injury without consistent evidence of direct infection of the vascular wall.
Circulation.
1999; 100(14): 1569–75. PubMed Abstract
| Publisher Full Text
- 217.
Ando K, Nakajima Y, Yamagishi T, et al.:
Development of proximal coronary arteries in quail embryonic heart: multiple capillaries penetrating the aortic sinus fuse to form main coronary trunk.
Circ Res.
2004; 94(3): 346–52. PubMed Abstract
| Publisher Full Text
- 218.
Baek ST, Tallquist MD:
Nf1 limits epicardial derivative expansion by regulating epithelial to mesenchymal transition and proliferation.
Development.
2012; 139(11): 2040–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 219.
Brewer CM, Majesky MW:
Branch Point Smooth Muscle Cells Highlighted by Novel Lineage Tracking Approach.
Circ Res.
2018; 122(2): 194–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 220.
Cooley BC, Nevado J, Mellad J, et al.:
TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling.
Sci Transl Med.
2014; 6(227): 227ra34. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 221.
Liu AC, Joag VR, Gotlieb AI:
The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology.
Am J Pathol.
2007; 171(5): 1407–18. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 222.
Majesky MW:
Development of coronary vessels.
Curr Top Dev Biol.
2004; 62: 225–59. PubMed Abstract
| Publisher Full Text
- 223.
Majesky MW:
Adventitia and perivascular cells.
Arterioscler Thromb Vasc Biol.
2015; 35(8): e31–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 224.
Majesky MW, Horita H, Ostriker A, et al.:
Differentiated Smooth Muscle Cells Generate a Subpopulation of Resident Vascular Progenitor Cells in the Adventitia Regulated by Klf4.
Circ Res.
2017; 120(2): 296–311. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 225.
Majesky MW, Schwartz SM:
An origin for smooth muscle cells from endothelium?
Circ Res.
1997; 80(4): 601–3. PubMed Abstract
- 226.
Roostalu U, Wong JK:
Arterial smooth muscle dynamics in development and repair.
Dev Biol.
2018; 435(2): 109–21. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 227.
Sawada H, Rateri DL, Moorleghen JJ, et al.:
Smooth Muscle Cells Derived From Second Heart Field and Cardiac Neural Crest Reside in Spatially Distinct Domains in the Media of the Ascending Aorta-Brief Report.
Arterioscler Thromb Vasc Biol.
2017; 37(9): 1722–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 228.
Souilhol C, Harmsen MC, Evans PC, et al.:
Endothelial-mesenchymal transition in atherosclerosis.
Cardiovasc Res.
2018; 114(4): 565–77. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 229.
Yoshida T, Gan Q, Franke AS, et al.:
Smooth and cardiac muscle-selective knock-out of Kruppel-like factor 4 causes postnatal death and growth retardation.
J Biol Chem.
2010; 285(27): 21175–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 230.
Wang JC, Bennett M:
Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence.
Circ Res.
2012; 111(2): 245–59. PubMed Abstract
| Publisher Full Text
- 231.
Desai P, Mencia-Trinchant N, Savenkov O, et al.:
Somatic mutations precede acute myeloid leukemia years before diagnosis.
Nat Med.
2018; 24(7): 1015–23. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 232.
Stemme S, Faber B, Holm J, et al.:
T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein.
Proc Natl Acad Sci U S A.
1995; 92(9): 3893–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 233.
Konstantinov IE:
In search of Alexander A. Maximow: the man behind the unitarian theory of hematopoiesis.
Perspect Biol Med.
2000; 43(2): 269–76. PubMed Abstract
| Publisher Full Text
- 234.
Maximov AA, Bloom W:
A Textbook of Histology. Philadelphia: W.B. Saunders. 1930. Reference Source
- 235.
Horvitz HR, Sulston JE:
"Joy of the worm".
Genetics.
1990; 126(2): 287–92. PubMed Abstract
| Free Full Text
- 236.
Garcia-Ojalvo J:
Cell Lineage Trees Bear Fruit.
Cell Syst.
2016; 3(6): 511–3. PubMed Abstract
| Publisher Full Text
- 237.
Silver GA:
Virchow, the heroic model in medicine: health policy by accolade.
Am J Public Health.
1987; 77(1): 82–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 238.
Waddington CH:
The strategy of the genes: a discussion of some aspects of theoretical biology. London: Allen & Unwin. 1957. Reference Source
- 239.
Trapnell C:
Defining cell types and states with single-cell genomics.
Genome Res.
2015; 25(10): 1491–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 240.
Link VM, Duttke SH, Chun HB, et al.:
Analysis of Genetically Diverse Macrophages Reveals Local and Domain-wide Mechanisms that Control Transcription Factor Binding and Function.
Cell.
2018; 173(7): 1796–1809.e17. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 241.
Fonseca GJ, Seidman JS, Glass CK:
Genome-Wide Approaches to Defining Macrophage Identity and Function.
Microbiol Spectr.
2016; 4(5). PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 242.
Gomez Perdiguero E, Klapproth K, Schulz C, et al.:
Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors.
Nature.
2015; 518(7540): 547–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 243.
Perdiguero EG, Geissmann F:
The development and maintenance of resident macrophages.
Nat Immunol.
2016; 17(1): 2–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 244.
Schulz C, Gomez Perdiguero E, Chorro L, et al.:
A lineage of myeloid cells independent of Myb and hematopoietic stem cells.
Science.
2012; 336(6077): 86–90. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 245.
Varol C, Landsman L, Fogg DK, et al.:
Monocytes give rise to mucosal, but not splenic, conventional dendritic cells.
J Exp Med.
2007; 204(1): 171–80. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 246.
Wlodarczyk A, Holtman IR, Krueger M, et al.:
A novel microglial subset plays a key role in myelinogenesis in developing brain.
EMBO J.
2017; 36(22): 3292–308. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 247.
Woollard KJ, Geissmann F:
Monocytes in atherosclerosis: subsets and functions.
Nat Rev Cardiol.
2010; 7(2): 77–86. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 248.
Moss NS, Benditt EP:
Spontaneous and experimentally induced arterial lesions. I. An ultrastructural survey of the normal chicken aorta.
Lab Invest.
1970; 22(2): 166–83. PubMed Abstract
- 249.
Dong J, Horvath S:
Understanding network concepts in modules.
BMC Syst Biol.
2007; 1: 24. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 250.
Langfelder P, Luo R, Oldham MC, et al.:
Is my network module preserved and reproducible?
PLoS Comput Biol.
2011; 7(1): e1001057. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 251.
Song L, Langfelder P, Horvath S:
Comparison of co-expression measures: mutual information, correlation, and model based indices.
BMC Bioinformatics.
2012; 13: 328. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 252.
Mason MJ, Fan G, Plath K, et al.:
Signed weighted gene co-expression network analysis of transcriptional regulation in murine embryonic stem cells.
BMC Genomics.
2009; 10: 327. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 253.
Subramanian A, Narayan R, Corsello SM, et al.:
A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles.
Cell.
2017; 171(6): 1437–1452.e17. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 254.
Aten JE, Fuller TF, Lusis AJ, et al.:
Using genetic markers to orient the edges in quantitative trait networks: the NEO software.
BMC Syst Biol.
2008; 2: 34. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 255.
Lee SI, Dudley AM, Drubin D, et al.:
Learning a prior on regulatory potential from eQTL data.
PLoS Genet.
2009; 5(1): e1000358. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 256.
Lee SI, Pe'er D, Dudley AM, et al.:
Identifying regulatory mechanisms using individual variation reveals key role for chromatin modification.
Proc Natl Acad Sci U S A.
2006; 103(38): 14062–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 257.
Li H, Chen H, Bao L, et al.:
Integrative genetic analysis of transcription modules: towards filling the gap between genetic loci and inherited traits.
Hum Mol Genet.
2006; 15(3): 481–92. PubMed Abstract
| Publisher Full Text
- 258.
Schwartz SM, Schwartz HT, Horvath S, et al.:
A systematic approach to multifactorial cardiovascular disease: causal analysis.
Arterioscler Thromb Vasc Biol.
2012; 32(12): 2821–35. PubMed Abstract
| Publisher Full Text
- 259.
Cao J, Packer JS, Ramani V, et al.:
Comprehensive single-cell transcriptional profiling of a multicellular organism.
Science.
2017; 357(6352): 661–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 260.
Cusanovich DA, Reddington JP, Garfield DA, et al.:
The cis-regulatory dynamics of embryonic development at single-cell resolution.
Nature.
2018; 555(7697): 538–42. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 261.
Drake TA, Schadt EE, Lusis AJ:
Integrating genetic and gene expression data: application to cardiovascular and metabolic traits in mice.
Mamm Genome.
2006; 17(6): 466–79. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 262.
Schadt EE:
Exploiting naturally occurring DNA variation and molecular profiling data to dissect disease and drug response traits.
Curr Opin Biotechnol.
2005; 16(6): 647–54. PubMed Abstract
| Publisher Full Text
- 263.
Schadt EE:
Association Studies: A genome-wide association approach to mapping the genetic determinants of the transcriptome in human populations.
Eur J Hum Genet.
2006; 14: 891–3. Publisher Full Text
- 264.
Campbell GR, Campbell JH, Manderson JA, et al.:
Arterial smooth muscle. A multifunctional mesenchymal cell.
Arch Pathol Lab Med.
1988; 112(10): 977–86. PubMed Abstract
- 265.
Chamley JH, Campbell GR, Burnstock G:
Dedifferentiation, redifferentiation and bundle formation of smooth muscle cells in tissue culture: the influence of cell number and nerve fibres.
J Embryol Exp Morphol.
1974; 32(2): 297–323. PubMed Abstract
- 266.
Du KL, Ip HS, Li J, et al.:
Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation.
Mol Cell Biol.
2003; 23(7): 2425–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 267.
Gan Q, Yoshida T, Li J, et al.:
Smooth muscle cells and myofibroblasts use distinct transcriptional mechanisms for smooth muscle alpha-actin expression.
Circ Res.
2007; 101(9): 883–92. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 268.
Hautmann MB, Adam PJ, Owens GK:
Similarities and differences in smooth muscle alpha-actin induction by TGF-beta in smooth muscle versus non-smooth muscle cells.
Arterioscler Thromb Vasc Biol.
1999; 19(9): 2049–58. PubMed Abstract
| Publisher Full Text
- 269.
Jin L, Yoshida T, Ho R, et al.:
The actin-associated protein Palladin is required for development of normal contractile properties of smooth muscle cells derived from embryoid bodies.
J Biol Chem.
2009; 284(4): 2121–30. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 270.
Kumar MS, Owens GK:
Combinatorial control of smooth muscle-specific gene expression.
Arterioscler Thromb Vasc Biol.
2003; 23(5): 737–47. PubMed Abstract
| Publisher Full Text
- 271.
Mack CP, Thompson MM, Lawrenz-Smith S, et al.:
Smooth muscle alpha-actin CArG elements coordinate formation of a smooth muscle cell-selective, serum response factor-containing activation complex.
Circ Res.
2000; 86(2): 221–32. PubMed Abstract
| Publisher Full Text
- 272.
Manabe I, Owens GK:
CArG elements control smooth muscle subtype-specific expression of smooth muscle myosin in vivo.
J Clin Invest.
2001; 107(7): 823–34. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 273.
Manabe I, Owens GK:
Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system.
Circ Res.
2001; 88(11): 1127–34. PubMed Abstract
| Publisher Full Text
- 274.
Owens GK:
Molecular control of vascular smooth muscle cell differentiation.
Acta Physiol Scand.
1998; 164(4): 623–35. PubMed Abstract
| Publisher Full Text
- 275.
Yoshida T, Owens GK:
Molecular determinants of vascular smooth muscle cell diversity.
Circ Res.
2005; 96(3): 280–91. PubMed Abstract
| Publisher Full Text
- 276.
Price DL, Porter KR:
The response of ventral horn neurons to axonal transection.
J Cell Biol.
1972; 53(1): 24–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 277.
Tapscott SJ, Lassar AB, Davis RL, et al.:
5-bromo-2'-deoxyuridine blocks myogenesis by extinguishing expression of MyoD1.
Science.
1989; 245(4917): 532–6. PubMed Abstract
| Publisher Full Text
- 278.
Bochaton-Piallat ML, Gabbiani F, Redard M, et al.:
Apoptosis participates in cellularity regulation during rat aortic intimal thickening.
Am J Pathol.
1995; 146(5): 1059–64. PubMed Abstract
| Free Full Text
- 279.
Guelfi S, Duffau H, Bauchet L, et al.:
Vascular Transdifferentiation in the CNS: A Focus on Neural and Glioblastoma Stem-Like Cells.
Stem Cells Int.
2016; 2016: 2759403. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 280.
Glass CK:
Genetic and genomic approaches to understanding macrophage identity and function.
Arterioscler Thromb Vasc Biol.
2015; 35(4): 755–62. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 281.
Belknap JK, Grieshaber NA, Schwartz PE, et al.:
Tropoelastin gene expression in individual vascular smooth muscle cells. Relationship to DNA synthesis during vascular development and after arterial injury.
Circ Res.
1996; 78(3): 388–94. PubMed Abstract
| Publisher Full Text
- 282.
Bergwerff M, DeRuiter MC, Poelmann RE, et al.:
Onset of elastogenesis and downregulation of smooth muscle actin as distinguishing phenomena in artery differentiation in the chick embryo.
Anat Embryol (Berl).
1996; 194(6): 545–57. PubMed Abstract
| Publisher Full Text
- 283.
Gadson PF Jr, Rossignol C, McCoy J, et al.:
Expression of elastin, smooth muscle alpha-actin, and c-jun as a function of the embryonic lineage of vascular smooth muscle cells.
In Vitro Cell Dev Biol Anim.
1993; 29A(10): 773–81. PubMed Abstract
| Publisher Full Text
- 284.
Lemire JM, Potter-Perigo S, Hall KL, et al.:
Distinct rat aortic smooth muscle cells differ in versican/PG-M expression.
Arterioscler Thromb Vasc Biol.
1996; 16(6): 821–9. PubMed Abstract
| Publisher Full Text
- 285.
Thyberg J:
Differentiated properties and proliferation of arterial smooth muscle cells in culture.
Int Rev Cytol.
1996; 169: 183–265. PubMed Abstract
| Publisher Full Text
- 286.
Red-Horse K, Ueno H, Weissman IL, et al.:
Coronary arteries form by developmental reprogramming of venous cells.
Nature.
2010; 464(7288): 549–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 287.
Marcu R, Choi YJ, Xue J, et al.:
Human Organ-Specific Endothelial Cell Heterogeneity.
iScience.
2018; 4: 20–35. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 288.
Nolan DJ, Ginsberg M, Israely E, et al.:
Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration.
Dev Cell.
2013; 26(2): 204–19. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 289.
Cunha GR, Hayward SW, Dahiya R, et al.:
Smooth muscle-epithelial interactions in normal and neoplastic prostatic development.
Acta Anat (Basel).
1996; 155(1): 63–72. PubMed Abstract
| Publisher Full Text
- 290.
Cunha GR, Young P, Hamamoto S, et al.:
Developmental response of adult mammary epithelial cells to various fetal and neonatal mesenchymes.
Epithelial Cell Biol.
1992; 1(3): 105–18. PubMed Abstract
- 291.
DiSandro MJ, Li Y, Baskin LS, et al.:
Mesenchymal-epithelial interactions in bladder smooth muscle development: Epithelial specificity.
J Urol.
1998; 160(3 Pt 2): 1040–6; discussion 1079. PubMed Abstract
| Publisher Full Text
- 292.
Caplice NM, Bunch TJ, Stalboerger PG, et al.:
Smooth muscle cells in human coronary atherosclerosis can originate from cells administered at marrow transplantation.
Proc Natl Acad Sci U S A.
2003; 100(8): 4754–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 293.
Sata M, Saiura A, Kunisato A, et al.:
Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis.
Nat Med.
2002; 8(4): 403–9. PubMed Abstract
| Publisher Full Text
- 294.
Bentzon JF, Weile C, Sondergaard CS, et al.:
Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice.
Arterioscler Thromb Vasc Biol.
2006; 26(12): 2696–702. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 295.
Holifield B, Helgason T, Jemelka S, et al.:
Differentiated vascular myocytes: are they involved in neointimal formation?
J Clin Invest.
1996; 97(3): 814–25. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 296.
Scott NA, Cipolla GD, Ross CE, et al.:
Identification of a potential role for the adventitia in vascular lesion formation after balloon overstretch injury of porcine coronary arteries.
Circulation.
1996; 93(12): 2178–87. PubMed Abstract
| Publisher Full Text
- 297.
Coen M, Gabbiani G, Bochaton-Piallat ML:
Myofibroblast-mediated adventitial remodeling: an underestimated player in arterial pathology.
Arterioscler Thromb Vasc Biol.
2011; 31(11): 2391–6. PubMed Abstract
| Publisher Full Text
- 298.
Davis J, Molkentin JD:
Myofibroblasts: Trust your heart and let fate decide.
J Mol Cell Cardiol.
2014; 70: 9–18. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 299.
Kaneishi NK, Cockerell CJ:
Histologic differentiation of desmoplastic melanoma from cicatrices.
Am J Dermatopathol.
1998; 20(2): 128–34. PubMed Abstract
| Publisher Full Text
- 300.
Otranto M, Sarrazy V, Bonté F, et al.:
The role of the myofibroblast in tumor stroma remodeling.
Cell Adh Migr.
2012; 6(3): 203–19. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 301.
Walker RA:
The complexities of breast cancer desmoplasia.
Breast Cancer Res.
2001; 3(3): 143–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 302.
Volz KS, Jacobs AH, Chen HI, et al.:
Pericytes are progenitors for coronary artery smooth muscle.
eLife.
2015; 4: pii: e10036. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 303.
Majesky MW, Dong XR, Hoglund V, et al.:
The adventitia: a progenitor cell niche for the vessel wall.
Cells Tissues Organs.
2012; 195(1–2): 73–81. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 304.
DeRuiter MC, Poelmann RE, VanMunsteren JC, et al.:
Embryonic endothelial cells transdifferentiate into mesenchymal cells expressing smooth muscle actins in vivo and in vitro.
Circ Res.
1997; 80(4): 444–51. PubMed Abstract
| Publisher Full Text
- 305.
Morimoto M, Liu Z, Cheng HT, et al.:
Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate.
J Cell Sci.
2010; 123(Pt 2): 213–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 306.
Suzuki T, Carrier EJ, Talati MH, et al.:
Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension.
Am J Physiol Lung Cell Mol Physiol.
2018; 314(1): L118–L126. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 307.
Nakajima Y, Mironov V, Yamagishi T, et al.:
Expression of smooth muscle alpha-actin in mesenchymal cells during formation of avian endocardial cushion tissue: a role for transforming growth factor beta3.
Dev Dyn.
1997; 209(3): 296–309. PubMed Abstract
| Publisher Full Text
- 308.
Chang L, Noseda M, Higginson M, et al.:
Differentiation of vascular smooth muscle cells from local precursors during embryonic and adult arteriogenesis requires Notch signaling.
Proc Natl Acad Sci U S A.
2012; 109(18): 6993–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 309.
Arribas SM, Briones AM, Bellingham C, et al.:
Heightened aberrant deposition of hard-wearing elastin in conduit arteries of prehypertensive SHR is associated with increased stiffness and inward remodeling.
Am J Physiol Heart Circ Physiol.
2008; 295(6): H2299–307. PubMed Abstract
| Publisher Full Text
- 310.
Wong LC, Langille BL:
Developmental remodeling of the internal elastic lamina of rabbit arteries: effect of blood flow.
Circ Res.
1996; 78(5): 799–805. PubMed Abstract
| Publisher Full Text
- 311.
Lu YW, Lowery AM, Sun LY, et al.:
Endothelial Myocyte Enhancer Factor 2c Inhibits Migration of Smooth Muscle Cells Through Fenestrations in the Internal Elastic Lamina.
Arterioscler Thromb Vasc Biol.
2017; 37(7): 1380–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 312.
Baeyens N, Bandyopadhyay C, Coon BG, et al.:
Endothelial fluid shear stress sensing in vascular health and disease.
J Clin Invest.
2016; 126(3): 821–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 313.
Rao RN, Falls DG, Gerrity RG, et al.:
Intimal thickness and layering, and smooth muscle cell phenotypes in aorta of youth.
Pathobiology.
2000; 68(1): 18–28. PubMed Abstract
| Publisher Full Text
- 314.
Williams KJ, Tabas I:
Lipoprotein retention--and clues for atheroma regression.
Arterioscler Thromb Vasc Biol.
2005; 25(8): 1536–40. PubMed Abstract
| Publisher Full Text
- 315.
Trigueros-Motos L, González-Granado JM, Cheung C, et al.:
Embryological-origin-dependent differences in homeobox expression in adult aorta: role in regional phenotypic variability and regulation of NF-κB activity.
Arterioscler Thromb Vasc Biol.
2013; 33(6): 1248–56. PubMed Abstract
| Publisher Full Text
- 316.
Jin H, Li DY, Chernogubova E, et al.:
Local Delivery of miR-21 Stabilizes Fibrous Caps in Vulnerable Atherosclerotic Lesions.
Mol Ther.
2018; 26(4): 1040–55. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 317.
Karnik SK, Brooke BS, Bayes-Genis A, et al.:
A critical role for elastin signaling in vascular morphogenesis and disease.
Development.
2003; 130(2): 411–23. PubMed Abstract
| Publisher Full Text
- 318.
Li DY, Faury G, Taylor DG, et al.:
Novel arterial pathology in mice and humans hemizygous for elastin.
J Clin Invest.
1998; 102(10): 1783–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 319.
Li DY, Brooke B, Davis EC, et al.:
Elastin is an essential determinant of arterial morphogenesis.
Nature.
1998; 393(6682): 276–80. PubMed Abstract
| Publisher Full Text
- 320.
Van Brussel I, Ammi R, Rombouts M, et al.:
Fluorescent activated cell sorting: an effective approach to study dendritic cell subsets in human atherosclerotic plaques.
J Immunol Methods.
2015; 417: 76–85. PubMed Abstract
| Publisher Full Text
Comments on this article Comments (0)