Keywords
Synthetic lethality, drug combinations, anticancer drugs, screening, synergy
Synthetic lethality, drug combinations, anticancer drugs, screening, synergy
More than six decades of concerted laboratory and clinical labor have enabled us to control and even cure some cancers1,2. Nonetheless, these successes represent the minority, and many tumors either are innately resistant or become resistant to the available therapies. We know that human cancer is complex with vast numbers of genetic alterations and it remains a major cause of human morbidity and death. In large part, we still fail to understand why some cancers are responsive to certain drugs whereas other tumors are not. This issue has become even more complicated because there has been an enormous increase in the number of US Food and Drug Administration (FDA)-approved treatments for cancer. Last year, Santos et al.3 divided the 154 FDA-approved cancer drugs broadly into four groups: 26 drugs are cytotoxic agents, such as those that disrupt DNA synthesis or integrity; 38 drugs are broadly cytotoxic and act at least partially through protein targets, such as the proteasome or microtubules; 85 drugs mechanistically target cancer-associated proteins; and 5 drugs act through unknown or non-protein targets. One should add to this list the two recent FDA approvals for chimeric antigen receptor–T cell (CAR–T) therapy to make a total of 156 agents (Figure 1). Combining multiple drugs to treat cancer is now standard clinical practice, but the actual components of the drug combinations often were generated empirically. The growth in the numbers of anticancer drugs makes this process much more challenging because of the number of possible theoretical combinations. Moreover, it is now believed that there are at least 600 cancer drivers across different cancers and there is enormous heterogeneity in the genetic composition of cells, even in a single tumor from a patient, not to mention among the metastases. Furthermore, tumor cells even in the same tumor exist in diverse environmental conditions, including differences in nutrient availability, pH, cytokine concentrations, oxygen levels, and reactive oxygen species quantities. This enormous complexity portends the development of even more anticancer agents2.
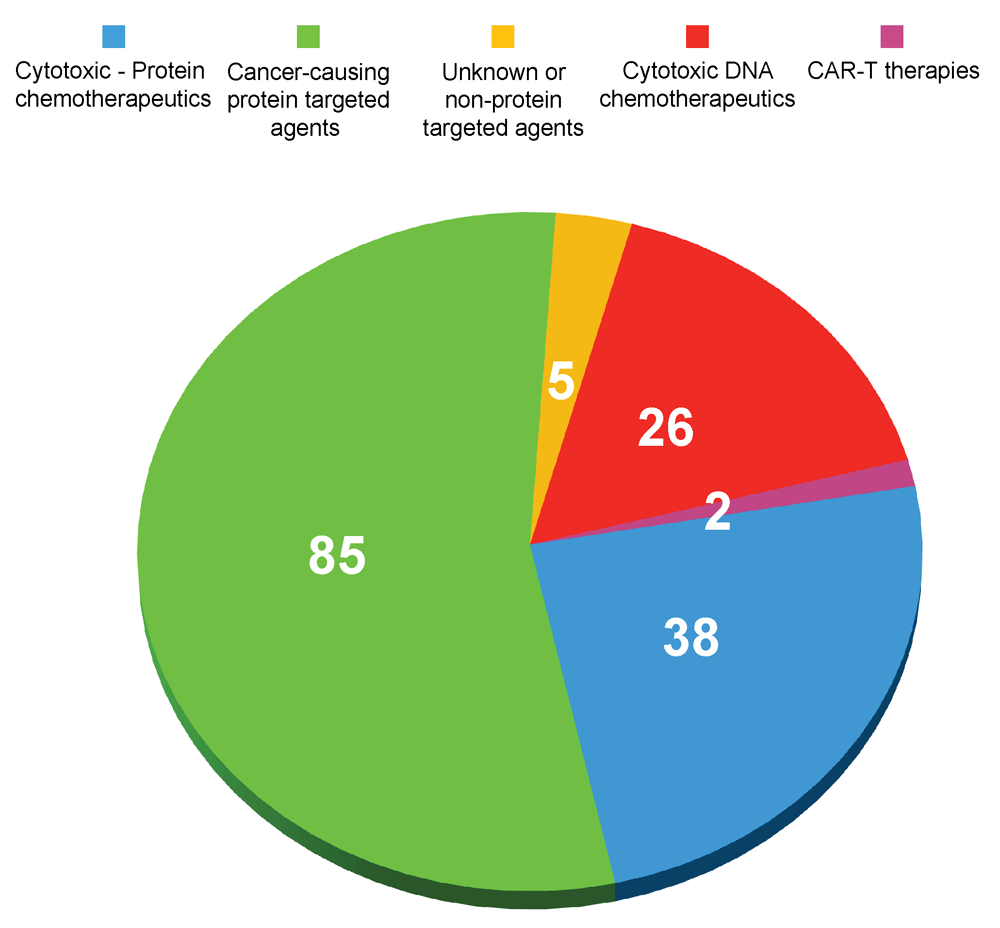
The classification scheme in this figure was inspired by Santos et al.3. CAR–T, chimeric antigen receptor–T cell.
Among the 600 cancer drivers, many, such as tumor suppressors, have been considered to be beyond the reach of traditional therapeutic intervention, but this concept is now being challenged1. This report examines one experimental approach that has recently become popular as a potential means to expose new druggable targets and to accelerate the identification of rational drug combinations: synthetic lethality screening. This somewhat mechanistically agnostic methodology enables one to uncover previously unknown cooperative interactions that sustain viability or another therapeutically relevant phenotype between the product of two genes, two compounds, or a compound and a gene product.
The concept of enhancing a therapeutic effect by combining different independent agents is ancient. Originally, combinations were purely observationally driven by perceptive and persistent medical practitioners. The principle of combination chemotherapy is currently most widely practiced and advanced in cancer. Historically, clinicians avoided using drugs that had overlapping organ toxicities and tried to apply drugs that had different molecular targets to reduce the potential for drug resistance. In the second half of the previous century, we developed a fairly good understanding about the biochemical reactions required for DNA synthesis, which enabled the creation of antimetabolites and antifolates, many of which we still use today. Thoughtful studies by Sartorelli et al.4,5, Darnowski and Handschumacher6, and Damon and Cadman7 emphasized the importance of targeting the rate-limiting steps in the biochemical reactions and identifying the optimal sequence of drug exposure. Mayer and Janoff8 illustrated the potential importance of maintaining constant ratios of the individual drugs whenever possible in the tumor and elsewhere.
The development of rigorous methods to quantify drug synergism has been an important area of early pharmacological and toxicological studies within and outside the area of cancer. One of the earliest published attempts to distinguish true compound synergy from additivity was published by Bliss9, who built his toxicological methodology on the pioneering mathematical work of J. H. Gaddum10. Since then, there have been multiple attempts to provide a universally accepted quantitative definition of drug synergy, but these have had only limited success. Chou highlighted the fact that as many as 13 different methods for determining synergism exist in the literature with considerable disagreement among them11. Reasons for the discrepancies stem from differing mechanistic assumptions, variable compound ratios, and the shapes of the individual drugs’ concentration response or dose response curves. A synergistic effect is not simply a result that is greater than the arithmetic sum of the effects of two drugs alone. For example, if a drug has a very steep dose response curve and one combines a minimally effective dose of compound A (a dose that caused a 1% effect or an ED1) with another minimally effective dose (ED1) of compound A, it is possible that an effect greater than an ED2 will be observed. This does not, however, indicate that compound A is acting synergistically with itself. A Cartesian plot of the relative effects of the individual compounds can provide an isobologram, which allows one to distinguish between additivity or synergy12. In cancer, one is generally interested in selective tumor cell killing, but many of the cytotoxic agents have low therapeutic indices and also damage non-malignant cells that are not involved in tumor growth or dissemination. Moreover, one should be striving for a large change rather than small changes in tumor viability, even with targeted therapies, if there is any hope for a meaningful survival advantage13. The mass-action law-based median-effect method has become one method that has been prominently employed in cancer studies11,14,15. Unfortunately, earlier attempts to mathematically define synergy or antagonism, including the median-effect method, assume idealized mass-action principles and employ logarithmic linearization data analyses, which can lead to poor model fitting16,17. Non-linear regression has been suggested to be a superior and more robust methodology to determine synergy16,17.
The fundamental concept of synthetic lethality generally has been attributed to the genetic studies conducted by Calvin Bridges with Drosophila melanogaster a century ago18. The actual term “synthetic lethality”, however, was coined 20 years later by Theodore Dobzhansky to describe the combination of two genetic events that created something new, in this case lethality, but only when both genes were lost19. It took almost half a century for cancer biologists and pharmacologists to embrace the idea of exploiting synthetic lethality to identify new drug combinations, initially using yeast and human cancer cells20,21. The growth of synthetic lethal screening and cancer has been reviewed elsewhere20–22. One popular pharmacologically oriented synthetic lethal cancer screening approach uses the expression of a gain-of-function proto-oncogene, such as an activated kinase, and evaluates a library of drugs seeking sensitization against a phenotypic endpoint, such as proliferation or death22. Alternatively, RNA interference (RNAi) or, more recently, clustered regularly interspaced short palindromic repeats (CRISPR)-based methods have been used to suppress the expression of a druggable gene product in cancer cells that are then treated with compounds from chemical libraries or RNAi libraries. Another strategy is to expose cancer cells to two different drugs and search for synergistic alterations in the phenotypic endpoint. Historically, gain-of-function mutations were viewed as much more druggable than loss-of-function mutations where restoration by adding a small molecule would be the goal1. Excitement for synthetic lethality studies was further stimulated by the concept of tumor cell “addiction” to oncogenes, formulated by the late Bernard Weinstein23, by which he meant cell death induced by disabling a required oncoprotein. Although the mechanistic basis for the so-called “addiction” was not well formulated, it had functional appeal for many investigators. The rapid realization that there was “addiction” even to non-oncogenic proteins opened up an even larger array of possible targets24. The use of large-scale small interfering RNA and short hairpin RNA screens revealed not only gain-of-function mutations that could be therapeutically approached yet also more challenging but, perhaps more interesting, loss-of-function mutations or deletions that are possible synthetic lethality participants25,26. Some of these synthetic lethality leads fell victim to the common pitfalls that often occur during preclinical cancer target validation, including the well-known off-target effects of small interfering RNA and short hairpin RNA13. Nonetheless, in 2014, the first drug identified by using synthetic lethality screen received FDA approval: the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib for the treatment of germline BRCA-mutated advanced ovarian cancer, which has cells that are DNA damage repair-deficient27,28. The use of the PARP inhibitor has now been extended to maintenance treatment for patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are having partial or complete responses to platinum-based chemotherapy. Thus, systematic mapping of disease drivers by using a synthetic letha lapproach can provide future therapeutic strategies by both identifying potential targets and highlighting key pathways that can be drugged.
In my opinion, the genetic and epigenetic heterogeneity and complexity within and between tumors are likely to dictate the continued exploitation of empirical approaches to discovering cancer drug combinations. Nonetheless, a recent publication provides an interesting window into how the future of drug combination studies might evolve29. The rapidly expanding genome editing tools, such as CRISPR-Cas930–32, provide powerful platforms, which can even be used in preclinical animal models. Manguso et al.29 used a pooled in vivo CRISPR-Cas9 strategy with transplantable B16 melanoma tumors injected into mice and then treated the mice with a monoclonal antibody against the PD-1 checkpoint and identified previously unknown genes that sensitized the tumors to the PD-1 immunotherapy in vivo. In particular, the authors found that suppression of the protein tyrosine phosphatase Ptpn2 increased the efficacy of anti-PD-1 therapy by enhancing interferon-gamma-mediated effects on antigen presentation and growth suppression. Although protein tyrosine phosphatases have historically been labeled “undruggable” at least with small molecules1, there have been several recent examples of successful drugging of this protein class33–36. This should encourage others to follow.
I expect that in vivo genomic editing screens will have a major role in defining the future of anticancer drug combinations. Some of the issues that exist with the current genomic editing methodology, however, are the permanence of the DNA changes; the quantal nature of the silencing; off-target endonuclease activity associated with the use of Cas9; and the possibility for transcriptional, translational, or post-translational compensation during the gene deletion steps. More transient editing methods that target RNA might help resolve concerns about the quantal and permanent nature of expression. Human tumors often have cells with amplified gene-encoding regions containing oncogenes or heterozygous loss of genes encoding tumor suppressors, which are challenging to emulate using current genomic editing technology. The timing or scheduling of the individual drug combinations could also have important consequences with respect to tumor response, and more reversible, concentration-controllable, methods could allow one to determine the optimal drug sequence and doses. Of course, the consequences of removing a protein are not always reproduced by the simple binding of a small molecule to the protein unless one is using methods, such as proteolysis targeting chimeras37, to deplete intracellular proteins. Another matter that is not easily addressed with any of the existing genomic editing methods is how to design combinations with multiple drugs.
Genetic studies have demonstrated that some synthetic lethal interactions are contextually dependent and cell type-specific22. For synthetic lethal interactions to be successfully transferred from laboratory models to humans, the contextual parameters will need to be carefully outlined in both fundamental and clinical studies. In the absence of well-defined and measurable conditions, a more general pan-synthetic lethal interaction might be more desirable than one that targets the so-called private interactions22. However, it should be noted that recent studies suggest that drug additivity or synergy may not be universally necessary for a positive benefit with drug combinations38.
Even with these limitations, there is considerable enthusiasm for the use of synthetic lethal strategies to accelerate the identification of anticancer drug combinations. Moreover, there appears to be no reason to believe that the synthetic lethality approach would not be generally useful for designing treatments for other non-malignant diseases. Nevertheless, a critical question remains as to whether synthetic lethal strategies, regardless of how they are designed, will actually accelerate the drugging of challenging targets or expose new clinically productive anticancer drug combinations. As we have learned repeatedly, cancer is a complex collection of diseases and appealing methodologies often do not live up to our expectations.
CRISPR, clustered regularly interspaced short palindromic repeats; FDA, US Food and Drug Administration; PARP, poly(ADP-ribose) polymerase; RNAi, RNA interference.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)