Keywords
Mediator subcomplex 12 (MED12), mutations, uterine leiomyomas, fibroadenomas of the breast, chronic lymphocytic leukemia (CLL), multiple tumors, DNA-RNA hybrids, Staphylococcus aureus
Mediator subcomplex 12 (MED12), mutations, uterine leiomyomas, fibroadenomas of the breast, chronic lymphocytic leukemia (CLL), multiple tumors, DNA-RNA hybrids, Staphylococcus aureus
First of all we would like to thank the three reviewers for considering our hypotheses on the molecular background of those uterine smooth muscle tumors and fibroepithelial tumors of the breast carrying mutations of Mediator Complex Subunit 12 ( MED12).
Compared to the first version we have addressed the criticisms and nearly all comments and suggestions raised by the reviewers. Also, an additional subheading entitled "Summary, weaknesses of the hypothesis, and conclusions" offers a critical synopsis mentioning open questions and weaknesses as e.g. the different possible mechanisms of transfer of the bacterial RNA into the putative target cell. Furthermore, Figures 2 and 3 (old numbers) have been condensed to one Figure (2, new number) to avoid redundancies leading to a reduced number of figures. On the other hand, by introducing subfigures to Figures 7 and 8 (new numbers each), additional information is provided on the description of a transcript comprising the putative hairpin sequence (Fig. 7B) and its similarity among different species of Staphylococcus (Fig. 8B).
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Surprisingly, the most common mutation in human tumors does not affect one of the famous suspects in the field (for review see (Vogelstein et al., 2013)) but the much less well-known gene encoding Mediator Complex Subunit 12 (MED12). MED12 is part of the transcriptional preinitiation complex CDK8 (Elmlund et al., 2006) and encoded by a gene that maps to the X-chromosome at Xq13.1. One obvious reason why its mutations so far have gained much less interest than those of other genes frequently mutated in human tumors is that they affect, to a large extent, benign tumors. Moreover, within malignant tumors, they are virtually absent from most of the predominant epithelial neoplasms like cancers of colon, breast, and lung (Kandoth et al., 2013) whereas they have been found at considerable frequencies in some non-epithelial malignant tumors having a possible origin from benign precursor lesions.
As to the benign tumors, however, mutations of MED12 occur as apparent driver mutations in a predominant subset of human uterine leiomyomas (Mäkinen et al., 2011; Markowski et al., 2012; McGuire et al., 2012), constituting the by far most frequent human symptomatic tumors of all. Likewise, these mutations are also found in a large subset of fibroadenomas of the breast (Lim et al., 2014; Piscuoglio et al., 2015; Yoshida et al., 2015), another frequent benign tumor which occurs predominantly in young and middle-aged women. Interestingly, they are not restricted to benign tumors but also frequently seen in their malignant counterparts, i.e. uterine leiomyosarcomas (Kämpjärvi et al., 2012; Markowski et al., 2013a; Pérot et al., 2012) and malignant phyllodes tumors (Nagasawa et al., 2015; Piscuoglio et al., 2015; Yoshida et al., 2015). Also, their presence in malignant tumors suggests that, albeit as a very rare event, certain additional mutations can trigger malignant transformation within formerly benign tumors harboring MED12 mutations. Apart from solid tumors, MED12 mutations recently were also detected in a significant percentage of roughly 5–9% of chronic lymphocytic leukemias (CLL) (Guièze et al., 2015; Kämpjärvi et al., 2015; Wu et al., 2017a). Furthermore, the same type of MED12 mutation was found in two canine vaginal leiomyomas (Markowski et al., 2013a).
A closer look at the MED12 mutations does not reveal apparent differences between the type of mutations when comparing the different tumor entities. While for CLL only a few cases have been reported so far, the mutations in uterine leiomyomas and fibrodadenomas of the breast predominantly are clustered in the 5´ region of exon 2 of the gene with only a few mutations affecting the intron 1-exon 2 boundary or, more rarely, exon 1 or the exon 1-intron 1 boundary. Most of them are single base exchanges clearly clustered at two nucleotides of codon 44 where, albeit with different frequencies, guanins are found to be replaced by either A, C, or T. Besides these single base replacements, deletions and, more rarely, indels, usually affecting exon 2 or the intron 1/exon 2 boundary are found which always leave the reading frame intact indicating that the mutations do not exert their tumor driving potential simply by abrogating the function of MED12. MED12 maps to the X-chromosome and, as revealed by cDNA sequencing, the mutations are apparently restricted to the active X-chromosome (Mäkinen et al., 2011; Markowski et al., 2012; McGuire et al., 2012).
We feel that for several reasons this highly frequent type of mutation might point to an unusual mechanism of mutagenesis underlying the development of the corresponding tumors. These reasons will be discussed herein and a hypothesis based on target-specific mutagenesis will be presented. Starting with a short introduction of the main tumors affected by MED12 mutations we will then address the molecular pathogenesis of uterine leiomyomas along with its clinical correlations followed by an in depth analysis of the pattern of MED12 mutations. Finally, we will present a hypothesis why these data indicate a so far unknown etiological mechanism favoring these particular highly frequent somatic alterations of the genome.
Besides a few very rare tumors, three main tumor entities are often affected by mutations of MED12. First, these three entities, i.e. uterine leiomyomas (fibroids), fibroadenomas of the breast, and chronic lymphocytic leukemias, will be introduced.
Uterine leiomyomas (UL) are benign smooth muscle tumors of myometrial origin with an apparently very low tendency to undergo malignant transformation. Depending on the location, the difference between submucosal, intramural, and subserosal UL can be distinguished. Roughly 40–70% of women in their reproductive age will develop UL with a well-documented higher prevalence among women of African and African-American origin. In this group, the UL develop on average at younger ages (Laughlin et al., 2010). In general, epidemiological studies have a revealed a lower risk of fibroid development associated with parity which may be related to tissue remodelling during pregancy (Baird & Dunson, 2003). Leiomyomas are often of large size and, as a matter of debate for more than 100 years, (Figure 1), multiple nodules occur at almost the same frequency or even more frequently than single nodules.
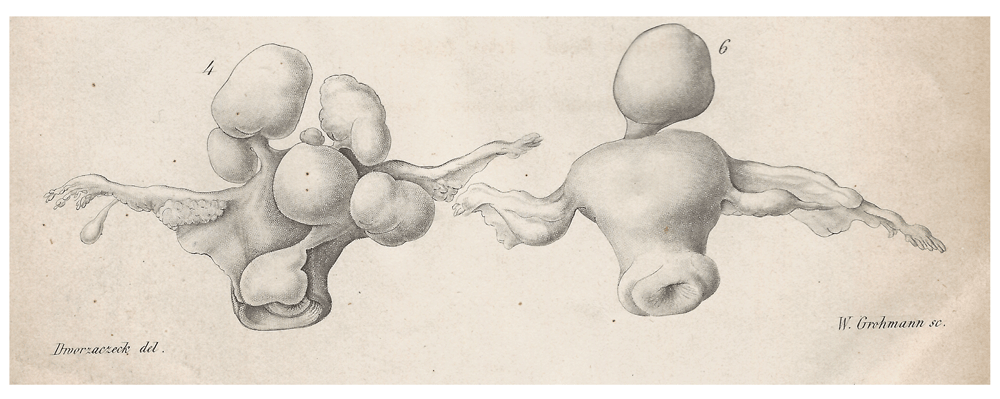
Illustration of a uterus carrying multiple leiomyomas of various sizes and location (4, left) and uterus with a large pedunculated subserous leiomyoma (right, 6). Copperplate engraving from Virchow´s series of lectures entitled “Die krankhaften Geschwülste” (“Morbid neoplasms”)(Hirschwald, Berlin, 1864/65).
Nevertheless, the majority of patients with UL are without symptom. The remaining 20–30% of the patients suffer from symptoms like in particular heavy menstrual bleeding, increased menstrual periods, reduced fertility, and pelvic pressure and pain. During menopause, UL cease growing and even shrink. Nevertheless, despite their benign nature UL are the most symptomatic human tumors of all and repeatedly have been reported even in mummies with the oldest of them dating back to the Middle Neolithic age (3,200–2,500 BC) (Fornaciari & Giuffra, 2012). Surgery (hysterectomy or myomectomy) is a common type of treatment, but a variety of other non-surgical methodologies to treat UL are available (for review see: (Williams, 2017)).
While the etiology of these frequent tumors remains unclear (McWilliams & Chennathukuzhi, 2017), a bit more is known about pathogenetic factors. As to basic principles of their molecular pathogenesis, UL behave like the vast majority of other benign and malignant tumors. This includes a monoclonal origin which is triggered by so-called driver mutations. In the case of multiple UL almost every single nodule has been found to be of independent clonal origin (Mashal et al., 1994). Accordingly, different nodules are usually characterized by different driver mutations in these cases. The role of myometrial stem cells as targets of driver mutations has been reviewed by Commandeur et al. (Commandeur et al., 2015).
Moreover, genetic subtypes, apparently belonging to different groups of driver mutations, exist. While MED12 mutations, as a group, constitute the most predominant genetic subtype (Mäkinen et al., 2011), another frequent subgroup of uterine leiomyomas carries rearrangements of the gene encoding the architectural transcription factor High Mobility AT-hook 2 (HMGA2), as a rule reflected by cytogenetically visible chromosomal translocations (Schoenmakers et al., 1995). Of note, the driver mutations of both subgroups occur in a mutually exclusive manner (Markowski et al., 2012). Besides these subgroups, other, more rare but also independent genetic subgroups of UL such as one characterized by either germline (hereditary leiomyomatosis and renal cell cancer (HLRCC), OMIM 605839) (Bayley et al., 2008) or somatic loss-of-function mutations of Fumarate Dehydrogenase (FH) seem to exist (Kämpjärvi et al., 2016).
Fibroadenomas of the breast are common benign tumors histologically composed of both stromal and epithelial components preferentially occurring in adolescent and young women. Their general incidence may be in the range of 10% in the corresponding age groups. Multiple tumors are not rare with some 10–15% of the patients having more than one FA.
Their name fibroadenomas (FA) and their classification as fibroepithelial tumors suggest a biphasic nature of these neoplasms. Nevertheless, as to their pathogenesis this classification seems to be misleading at least in the majority of cases because mutations are restricted to the stromal component of the tumors (Mishima et al., 2015). Between 50% and 60% of the FA harbor MED12 mutations (Lim et al., 2014; Mishima et al., 2015; Pfarr et al., 2015) Histologically, the occurrence of MED12 mutations correlates highly significant with the so-called intracanalicular growth pattern (Mishima et al., 2015; Pfarr et al., 2015). Interestingly, MED12 mutations have also been found in considerable percentages of other breast tumors of presumed stromal origin Phyllodes tumors and malignant Phyllodes tumors. In these tumors, the types of MED12 mutations are not obviously discernible from those observed in UL and FA.
In Western countries, chronic lymphocytic leukemia (CLL) is the most common type of leukemia in adults. The American Cancer Society expects an estimated number of about 20,940 new cases of CLL in the United States in 2018 with about 4,510 CLL-related deaths. Overall, CLL accounts for about one-quarter of the new cases of leukemia. The average person's lifetime risk of getting CLL is about one in 175 (0.57%). The average age at the time of diagnosis is around 70 years, with men slightly more often affected than women. Frequently, the disease is detected in patients not yet showing any severe symptoms and in its early stages patients often undergo a ‘watch and wait’ period prior to starting therapy.
As to its pathogenesis CLL is a monoclonal leukemia of B-cell origin with a number of subsets that can be distinguished based on their genetic characterization, apparently pointing to different driver mutations. The genetic alterations also allow the stratification of groups which may require different therapeutic approaches. As a valid predictive parameter, deletions and mutations of the TP53 gene are associated with a worse prognosis than other types of genetic changes and influence therapeutic decisions. Recently, mutations of MED12 have been added to the list of potential driver mutations in CLL. Across the sub-types, they constitute a relatively rare genetic group affecting roughly 5–9% of CLL patients (Guièze et al., 2015; Kämpjärvi et al., 2015; Wu et al., 2017a). Kämpjärvi et al. (Kämpjärvi et al., 2015) have presented evidence that MED12 mutations may represent a marker of worse prognosis.
According to the high prevalence of uterine leiomyomas MED12 mutations are by far best investigated in this tumor type. Thus, we have now characterized the subset UL affected and described how they can be distinguished from other types of UL.
There is ample evidence that HMGA2 rearrangements and MED12 mutations occur mutually exclusively in UL and thus constitute independent driver mutations (Markowski et al., 2012). Similarly, somatic MED12 mutations and biallelic Fumarate Hydratase (FH) inactivation occur in mutually exclusive manner in both HLRCC syndrome-associated and sporadic uterine leiomyomas suggesting that the latter constitutes a third small group with an independent molecular pathogenesis (Kämpjärvi et al., 2016). Accordingly, each of these genetic alterations alone as a driver mutation seems to be sufficient to induce the development of an UL without requiring any further mutations.
As to rearrangements of HMGA2 and mutations of MED12, the transcriptome of tumors of both groups clearly differs with MED12 mutation–positive and HMGA2-overexpressing samples clustering in distinct branches (Mehine et al., 2013). Accordingly, both mutations allow the two major genetic subtypes of UL to be distinguished, and the question arises whether or not the genetic subtypes are also reflected by a different clinical behavior and histopathology.
Tumors carrying HMGA2 rearrangements are usually solitary and, on average, of larger size than those with MED12 mutations, also usually presenting as single tumors (Markowski et al., 2014a) whereas the latter are smaller and often co-occur with other clonally independent nodules of the same genetic type (Mäkinen et al., 2011; Markowski et al., 2012; Markowski et al., 2014a). Among the women affected by MED12-mutation positive UL, more than two-thirds had more than one nodule (Heinonen et al., 2017) (Figure 2A) of this type. Even more impressive, in the same study 52 (8.7%) MED12-mutation positive tumors made their appearance as the sole tumor with this mutation, while 547 (91.3%) tumors were associated with at least one other tumor of this genetic subgroup (Figure 2B).
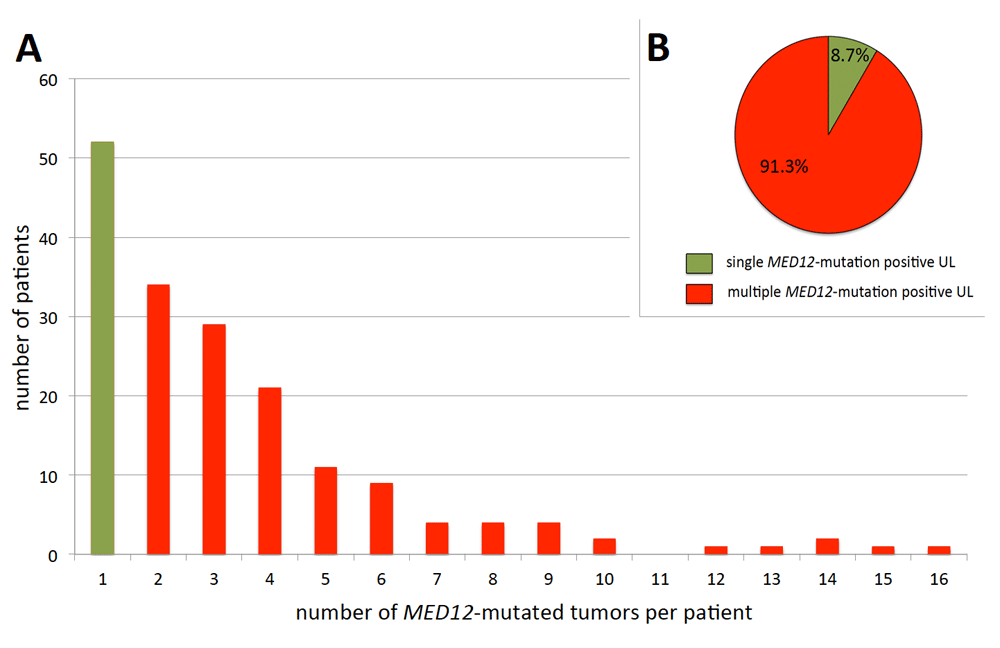
Abscissa: number of UL/patient, ordinate: number of patients in the corresponding category. For this diagram data on MED12 alterations published by Heinonen et al. (Heinonen et al., 2017) have been used. Overall, 52/176 women carried a single MED12-mutated UL compared to 124/176 with more than one such tumor (A) and proportion of MED12-mutated UL appearing as single tumor (52/599) vs. those accompanied by at least one other MED12-mutated UL (547/599) (B). Data according to Heinonen et al. (2017).
To explain the high frequency of MED12 mutated tumors among multiple leiomyomas, Heinonen et al. speculated that "the multiplicity of MED12-mutation-positive leiomyomas may derive from genetic predisposition and/or environmental factors rendering the myometrium susceptible to selection for MED12 mutations" (Heinonen et al., 2017). However, as outlined later herein, the association of multiple tumors with MED12 mutations may be a key to the etiology of this type of UL.
Furthermore, MED12-mutated UL are also significantly associated with a subserous location compared to UL lacking this mutation (Heinonen et al., 2017). As to histopathological features, a recent study by Wu et al. revealed that approximately 90% of the cells in HMGA2-rearranged UL were smooth muscle cells showing an overexpression of the protein, while in MED12-mutated UL a similar number of smooth muscle cells and other cells, i.e. mostly tumor-associated fibroblasts, were detected. These latter fibroblasts were lacking MED12-mutations (Wu et al., 2017b) and thus apparently can be classified as by-stander cells. This fits with an earlier observation that in cell cultures of leiomyomas with MED12-mutations a rapid disappearance of mutated cells was seen that became replaced by wild-type cells, thus challenging the results of a variety of in vitro experiments on the biology of UL (Bloch et al., 2017; Markowski et al., 2014b).
Besides UL, the occurrence of MED12 mutations has been well-documented in malignant uterine smooth muscle tumors (leiomyosarcomas) and smooth muscle tumors of uncertain malignant potential (STUMP), too (Holzmann et al., 2015; Pérot et al., 2012). Hence, an origin of these tumors from pre-existing UL has been suggested. In contrast, similar cases with HMGA2 rearrangements have not been reported yet.
To gain further insight into the biology of MED12-mutated UL, Heinonen et al. have undertaken a systematic attempt to check all feasible distinct tumors with a size of 1 cm or larger in diameter from hysterectomy uteri for MED12 mutations (Heinonen et al., 2017). In their study, 599 out of 763 leiomyomas carried MED12 mutations (79%). Next, the data provided by the study of Heinonen et al. (Heinonen et al., 2017) have been used to analyze the number of MED12-mutation positive UL per patient. While it was shown before that in the majority of patients having surgery MED12-mutated tumors do not make their appearance as single nodules but instead are accompanied by other yet clonally independent tumors of this same genetic type (cf. Figure 2), we were also interested to see how the number of tumors per patient is distributed in this genetically distinct group of tumors. Obviously, a slow decrease of the number of MED12-mutation positive tumors is noted (Figure 2A). Nevertheless, from these figures it is not possible to draw conclusions on the overall frequency and distribution of MED12-positive UL in the population because the results are biased by their restriction to symptomatic patients who had undergone surgical treatment. Nevertheless, they correspond more or less to the tumor numbers in general as seen from numerous other studies and thus the question arises if the MED12-positive tumors can be distinguished from the remaining UL as suggested by previous estimations (cf. Figure 2). Thus, we have next investigated if, in the case of multiple tumors, the percentage of MED12-mutation positive UL remains constant independent of the total number of tumors per patient. Surprisingly, it was noted that with a growing number of tumors, the percentage of MED12-mutated tumors clearly increased. Among solitary leiomyomas, only less than 40% of the tumors carried MED12 mutations but their frequency was approaching nearly 100% if twelve or more tumors were present per patient (Figure 3). Thus, in contrast to other mutations, those of MED12 seem to become more likely with an increase of the number of tumors.
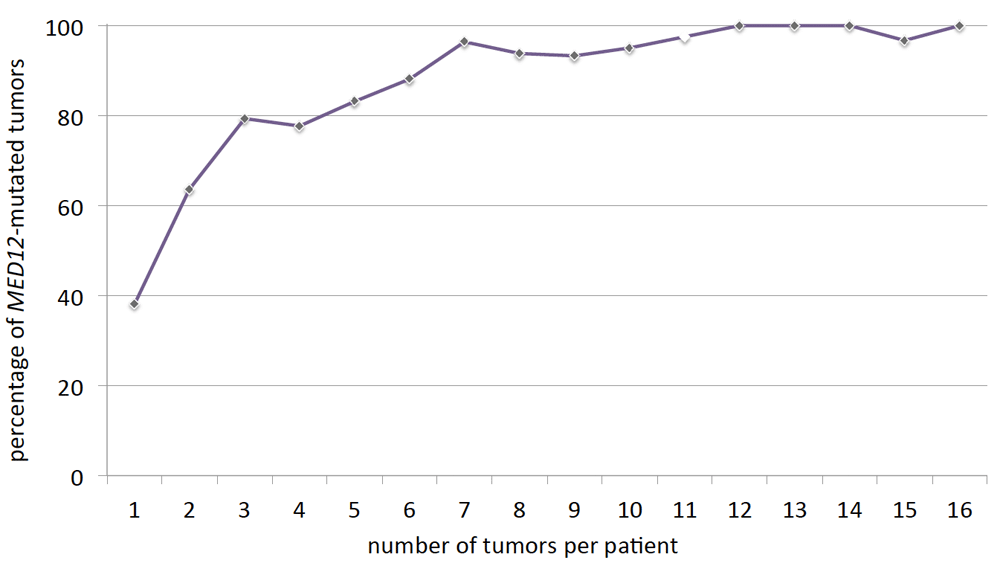
Increasing percentage of MED12-mutation positive uterine leiomyomas (ordinate) with the number of tumors per patient (abscissa). Open rhombus indicates an interpolated value because no patients with 11 UL were present. For this diagram data on MED12 alterations published by Heinonen et al. (2017) have been used.
Along with previous data this distribution confirms that the occurrence of multiple leiomyomas nearby can be exclusively attributed to just one genetic mechanism, i.e. MED12 mutations. For example, in a study by Markowski et al. only 26/179 (14.5%) of MED12-mutated UL were single tumors while the corresponding number for HMGA2-rearranged UL was 14/20 (70%). In none of the latter cases, a HMGA2-rearranged UL was accompanied by another one with HMGA2 rearrangement (Markowski et al., 2014a). This contradicts a statement by Mehine et al. that a shared clonal origin as a common feature of leiomyomas not carrying MED12 mutations offers one explanation for the common occurrence of multiple concurrent lesions (Mehine et al., 2015). Instead, a multitude of fibroids mainly appears to be a problem almost exclusively restricted to MED12-mutation positive tumors and we thus decided to analyze and compare the different MED12 mutations in more detail.
MED12 mutations occur in uterine smooth muscle tumors, fibroepithelial tumors of the breast and in chronic lymphocytic leukemia. As a rule, only subsets display these genetic abnormalities which range from the predominant alterations in UL to those detected only in a small percentage of cases in CLL. So far, there is no evidence that the tumors affected by MED12 mutations in the three tumor entities described above differ with respect to their types of mutations. It has been speculated that unknown factors favor the occurrence of these particular mutations. To get further ideas on factors favoring them, the patterns of mutations have now been analyzed in more detail and compared between the different tumor entities paying particular attention to the small deletions occurring in exon 2 or at the intron 1-exon 2 boundary.
In most cases single nucleotide exchanges are found with a clear predominance of those affecting nucleotides 130 and 131 belonging to codon 44. Less frequently other codons are mutated. Besides single nucleotide exchanges, deletions of small segments of the gene with varying sizes as well as indels affecting exon 2 or the intron1-exon 2 boundary are seen in some cases. As a rule, however, the transcript though affected by the deletions remains in frame. As to these latter genomic alterations accounting for roughly 15% of MED12 mutations, we have analyzed the positions of the deleted bases from a variety of papers analyzing UL, fibroepithelial tumors, and CLL. Adding the number of deleted bases per each position reveals an almost symmetric distribution that is clustered around the hotspot of single nucleotide exchanges (Figure 4). Accordingly, e.g. in the Heinonen et al. series, deletions encompassing nucleotides 129–134 or part of them are clearly more frequent than those outside this fragment. This clustering becomes evident when analyzing uterine smooth muscle tumors and fibroepithelial tumors of the breast alone, whereas in case of CLL only a few cases have been reported in the literature so far. In a previous study by our group, the beginning of the MED12 deletions observed in uterine smooth muscle tumors was mostly located within exon 2 but in rare cases also upstream of the splice site within intron 1. Their size mainly ranged between 3 and 36 bp with a clear predominance of 15 and 18 bp (Markowski et al., 2013b). Of note, an analysis of the data provided by Heinonen et al. for UL revealed that as very rare exceptions even larger deletions as well as those residing in exon 1 can occur.
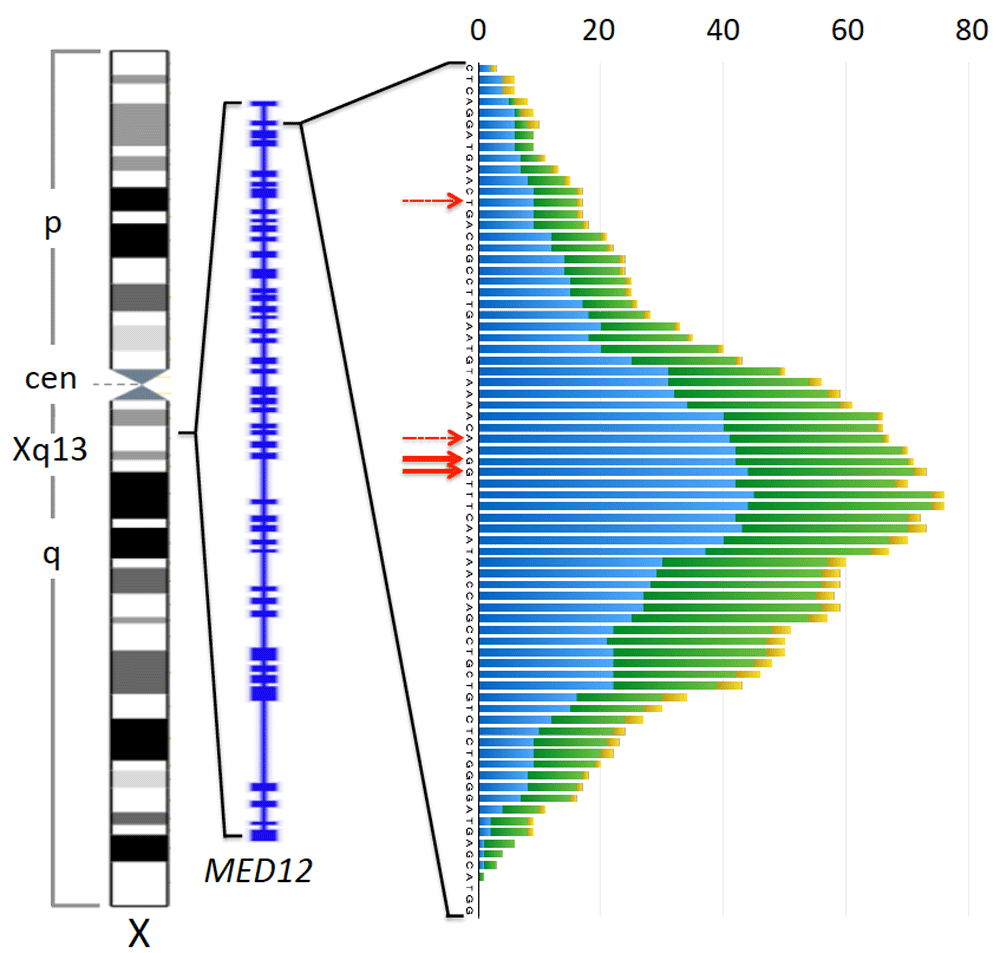
Left to right: Ideogram of the X-chromosome (commons.wikimedia.org), exon-intron structure of MED12 (NCBI map viewer), and plot depicting frequency of deletions at each position around the preferred site of single nucleotide exchanges (red solid arrows) seen in uterine smooth muscle tumors (blue), fibroepithelial tumors of the breast (green), and chronic lymphocytic leukemia (yellow). Deletions are plotted across all deleted base positions. Minor preferred sites of single nucleotid exchanges within exon 2 are indicated by dashed red arrows. For this diagram data on MED12 deletions from the following articles have been used: (Guièze et al., 2015; Kämpjärvi et al., 2015; Lim et al., 2014; Markowski et al., 2013b; Mishima et al., 2015; Nagasawa et al., 2015; Ng et al., 2015; Pfarr et al., 2015; Yoshida et al., 2015) only those deletions beginning and ending in the displayed region have been considered.
As to size and position of these deletions, there is also no obvious difference between UL, fibroepithelial tumors of the breast, and CLL (Figure 4). Overall, this pattern of small deletions of various sizes clustered around the hotspot of single nucleotide exchanges in general could be explained by bias caused by a higher proliferative activity of UL characterized by deletions encompassing the central hotspot area which accordingly would be of larger size and more likely to become symptomatic. Nevertheless, according to our evaluation of the Heinonen series Heinonen et al. (2017), the average size of UL with deletions encompassing any of the nucleotides 129–134 (average: 3.32) does not significantly differ from those having deletions outside this region (average: 3.35). Thus, such an obvious explanation seems less likely to explain the distribution which, however, resembles the results of genome editing based on targeted double-stranded breaks as for example those resulting from the usage of the CRISPR/Cas9 system (e.g. cf. Paquet et al., 2016).
If these mutations indeed arise by certain types of repair of site-specific DNA changes, one might expect that many other mutations occur in the target region of MED12. Of these, only the “active ones”, i.e. those leaving the open reading frame intact and driving tumorigenesis, will lead to a clonal proliferation of their target cell giving rise to an UL whereas cells with other mutations of the hotspot region will remain quiescent or even become apoptotic (Figure 5). Therefore, future studies aimed at the detection of these “non-driving” mutations in single cells, especially from patients suffering from a multitude of UL, may be a reasonable attempt. However, from the pattern of nucleotide exchanges and deletions, a commonly affected sequence can be depicted that may be related to the etiology of UL.

Model illustrating the occurrence and selection of MED12 mutations during the course of leiomyoma development. The scheme suggests that of a larger number of MED12-mutations only those associated with a gain of function act as driver mutations giving rise to UL.
MED12 mutations constitute highly frequent driver mutations in uterine leiomyomas and fibroadenomas, i.e. two tumor entities that occur almost exclusively in middle-aged and young women, respectively. In uterine leiomyomas, they even represent the by far most frequent genetic subtype with a clearly preferential occurrence in the case of multiple tumors. This is in sharp contrast to the other main genetic subtype of UL characterized by rearrangements of HMGA2 usually making its appearance in solitary nodules not accompanied by other tumors of the same genetic subtype.
It seems difficult to explain these findings just by independent random mutations followed by their selection. Nevertheless, additional factors favoring this multitude of tumors with independent clonal origin carrying the same type of mutation have remained enigmatic. After myomectomy, such factors may also account for the risk of recurrences that clearly increases with the number of UL that had been removed (Doridot et al., 2001; Fauconnier et al., 2000). Heinonen et al. (2017) have speculated that either genetic predisposition or environmental factors rendering the myometrium susceptible to selection for MED12 mutations may contribute to the multiplicity of MED12-mutation positive tumors. To describe the development of multiple tumors, those two explanations are well-compatible with a model of clonally unrelated nodules that occur successively and are endowed with a different growth rate as depicted in Figure 6A. Another alternative explaining the multiplicity of UL is the occurrence of clonally related nodules with marked genetic evolution as shown by Mehine et al. (Mehine et al., 2015) for UL lacking MED12 mutations and depicted here as Figure 6B. Nevertheless, a variety of studies indicate that most tumors with MED12 mutations are not clonally related and the multiplicity of these lesions thus needs other explanations. In addition to these models, the potential roles of infectious agents warrant consideration. The infection may lead to a synchronous initiation of multiple clonally independent lesions endowed with a different growth potential (Figure 6C).
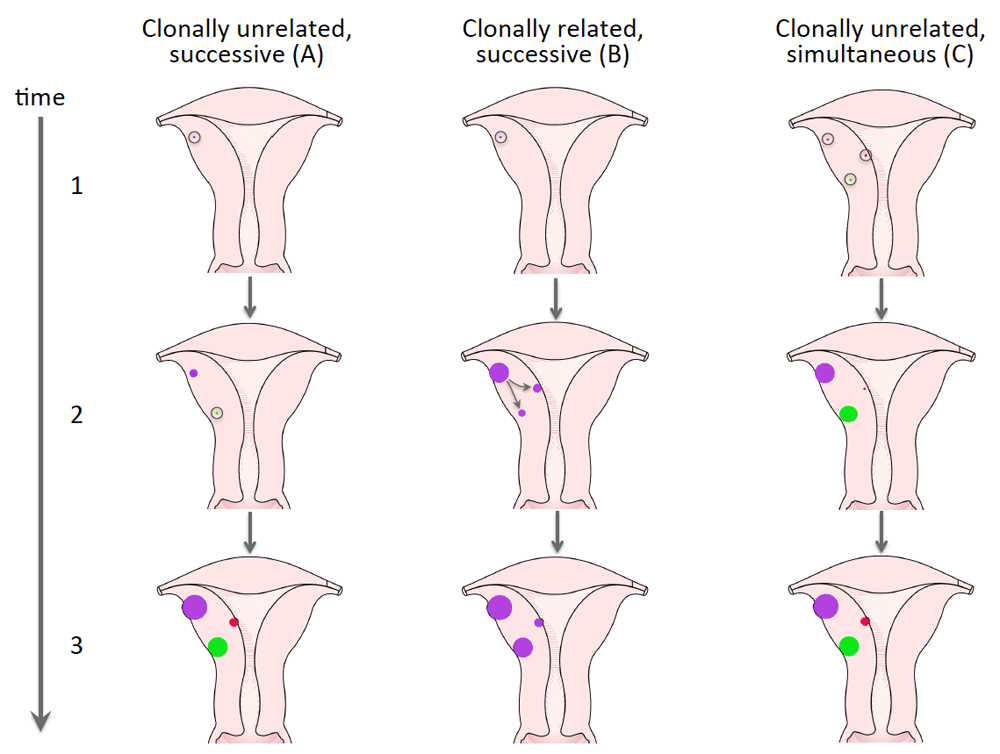
Three alternative models explaining the development of multiple uterine leiomyomas are depicted: (A) corresponds to a prima facie model of myomagenesis with multiple independently developing nodules having a different growth rate. (B) Illustrates the development of multiple clonally related tumors from one common predecessor as described by Mehine et al. (Mehine et al., 2015) for MED12-mutation negative UL. (C) offers an alternative explanation for the growth of multiple clonally unrelated nodules with simultaneous initiation but a different growth rate. Open circles indicate single mutated cells of origin affected by driver mutation.
As possible factors certain types of bacteria, e.g. those involved in reproductive tract infections (RTIs), have long been suggested to be a cause of UL (e.g. (Witherspoon & Butler, 1934)). In the United States both reportable RTIs (i.e. chlamydia and gonorrhea) and fibroids disproportionately burden African American women which lead to the conclusion that the growth of fibroids might be triggered by inflammatory infections associated with the RTIs. Nevertheless, when exploring the relationship between self-reported RTIs and fibroid size, number, and total volume Moore et al. did not find strong associations (Moore et al., 2015). In a recent contribution by the same group women seropositive for genital Chlamydia trachomatis were even found to be less likely to have fibroids (Moore et al., 2018). In line with these findings, in the study by Heinonen et al. neither a history of pelvic inflammatory disease (PID) nor of Chlamydia infection was found to be significantly associated with the MED12-type UL while PID turned out to be significantly associated with the occurrence of MED12-wild type UL (p 0.0024) (Heinonen et al., 2017).
Akin to bacteria, viruses have also been suggested to be involved in the development of UL (Bullerdiek, 1999). For example, EBV is known as a factor associated with the development of extra uterine smooth muscle tumors in HIV and post-transplant patients (see e.g. (Miettinen, 2014; Purgina et al., 2011; Ramdial et al., 2011)). However, so far no association between EBV and uterine leiomyomas has been demonstrated. As to another virus of the Herpes group, a recent study failed to reveal a significant association between HSV-2 seropositivity and the presence of fibroids (Moore et al., 2016) and in general no convincing evidence for involvement of viruses in the pathogenesis of UL has been presented.
While infectious diseases as etiological agents of UL repeatedly have been assumed as such the question arises as to how they could act by site-specific targeting the hotspot region of mutations residing within exon 2 of MED12 and which infectious agents are possible candidates. We will herein present the opinion that the interaction between the human DNA and foreign nucleic acids derived from the infection plays a causal role. As an unorthodox hypothesis stimulating further discussions, we would like to advance the hypothesis that the MED12 mutations result from cleavage of R-loop structures. By definition, R-loops are derived from double stranded DNA where one strand forms a stable DNA-RNA hybrid helix whereas the former associated DNA strand remains single-stranded. R-loops with an “exposed” stretch of single-stranded DNA can give rise to instability and DNA double-strand breaks (Aguilera & Gómez-González, 2017; Freudenreich, 2018; Su & Freudenreich, 2017). While the hybrid helix is usually composed of DNA with endogenous RNAs it seems possible that such helices can be formed with foreign RNA as well. To this end, it has been hypothesized that circulating exogenous RNA sequences after their uptake may influence the function of cells through miRNA-like mechanisms (Wang et al., 2012) suggesting direct influences of these sequences of the microbiome on its host´s cells.
To search for possible sequence homologies we have depicted a target region 5´TGTAAAACAAGGTTTCAATAAC3´ covering 10 nucleotides upstream and downstream each of the two c. 130 and c. 131 (GG), respectively (cf. Figure 5). Of the resulting list with at least 15 identical nucleotides, a variety of human pathogens have been identified as e.g. Bacteroides fragilis (nt 2-20), Klebsiella pneumoniae (nt 2-20), Escherichia coli (nt 3-20), Vibrio vulnificus (nt 6-22), Staphylococcus aureus (nt 6-22, and nt 1-16, respectively), Staphylococcus argenteus (nt 6-22), and Clostridium botulinum (nt 3-19). When searching for abundantly expressed sequences an interesting candidate emerged. A 16-base pair sequence identical to the sense strand of the sequence of a Staphylococcus aureus 27-tRNA gene cluster immediately 3’ to an rRNA operon (Green & Vold, 1993) was noted (Figure 7A). A transcript containing this sequence has been described (Tan et al., 2015, GeneBank accession GBKB01001045, Figure 7B). The homology covers a palindromic sequence which may act as a terminator of transcription (Green & Vold, 1993) and may also lead to the formation of a hairpin structure stabilizing the RNA molecule (Figure 8A). Of note, very similar palindromic sequences have been found at the same position of the genomes of other species of Staphylococcus (Figure 8B). Nevertheless, whereas the palindromic arms have high similarity the central part, the three AAA have undergone transversion to TTT. Are these molecules likely to exist as circulating RNAs? Staphylococcus aureus belongs to the phylum of Firmicutes in general constituting the third most abundant sequence population in human plasma with a significant number of the reads mapping to various bacterial ribosomal RNAs and tRNAs (Wang et al., 2012). More specifically, evidence has been presented that RNA species from Staphylococcus are commonly present in blood (Leung & Wu, 2015). Of note, Firmicutes are not rarely found to be part of the microbiota of the female genital tract with Staphylococcus sp. e.g. being the most abundant genus recovered from the fallopian tubes (Pelzer et al., 2018). This presence is not necessarily supposed to result from an ascending infection since e.g. hematogenous spread as well has been documented to be the reason for bacterial colonization in other mammals and may contribute to the genital tract microbiota (for review see Baker et al. (2018). In general, Staphylococcus species have been identified not only as part of the uterine microbiota (Chen et al., 2017; for review see Baker et al., 2018) but also of pancreatic and breast (healthy as well as benign and malignant disease) microbiota (Thomas et al., 2018; Urbaniak et al., 2016). Accordingly, the mere presence of Staphylococcus can be supposed not to be associated with bacteremia in most women. Interestingly, in HeLa cells Staphylococcus epidermidis isolated from breast cancer patients was found to be able to induce DNA double-stranded breaks (Urbaniak et al., 2016).
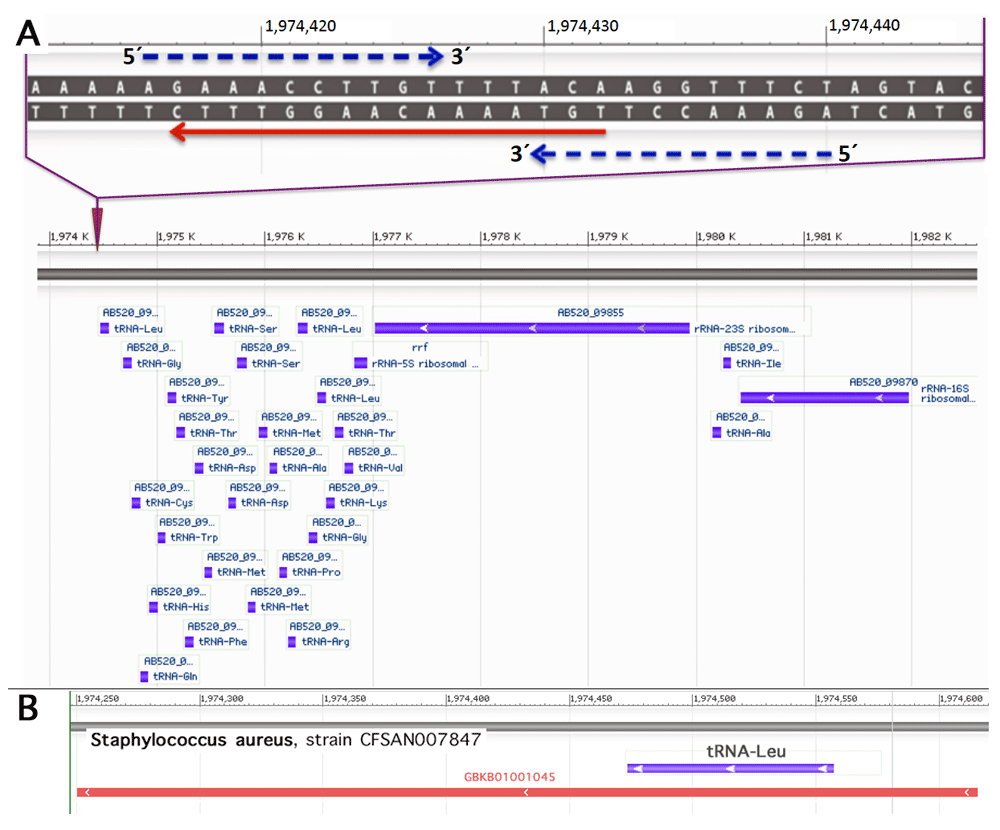
Upper part: Homology of the human MED12 hotspot region (red arrow) with the sense strand of the sequence of the Staphylococcus aureus 27-tRNA gene cluster immediately 3’ to a rRNA operon (Green & Vold, 1993). Blue dashed arrows indicate a palindromic sequence. Sequence from Staphylococcus aureus strain CFSAN007847 chromosome, complete genome; GenBank: CP017684.1; GenBank: FASTA; NCBI Blast. Lower part: bacterial tRNA and rRNA genes of the operon adjacent to the site of homology are shown in blue (A). Transcript containing parts of the of the operon and its 3´ vicinity including the homologous region (Tan et al., 2015: GeneBank accession GBKB01001045)(B).
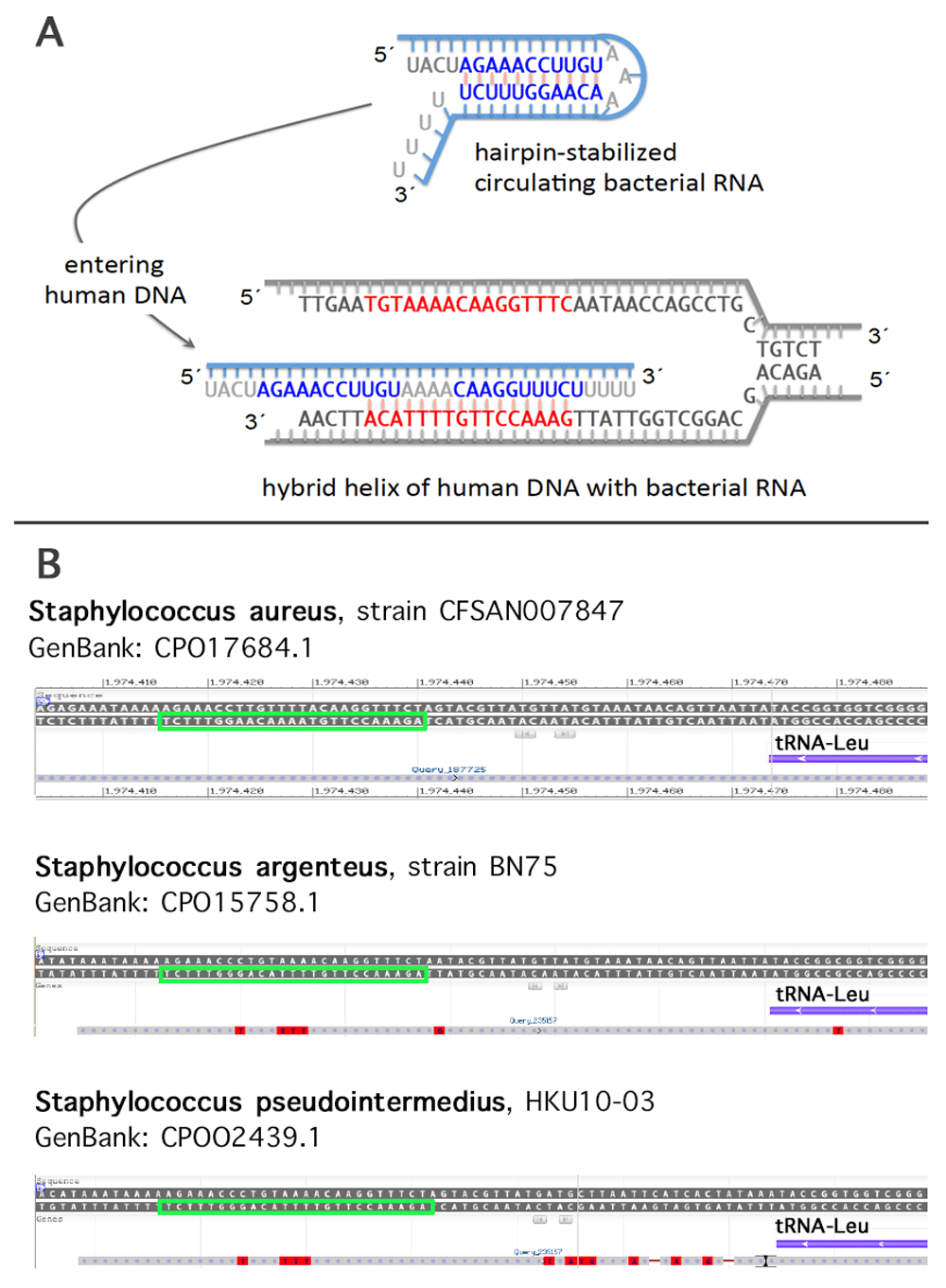
As an example for the putative sequence of the human microbiome inducing site specific mutations this scheme refers to the sequence depicted in Figure 7 (A). A similar palindromic structure is present at the same position of some other Staphylococcus species as shown here as examples for S. argenteus and S. pseudointermedius, respectively (green squares). (B)
We have addressed here the genetically distinct group of UL showing mutations of MED12. These mutations apparently act as the drivers in the majority of UL particularly occurring in women having multiple nodules and make UL in general the most frequent symptomatic human tumors at all. Nevertheless, in almost all cases these nodules appear to be of independent origin rather than clonally related. As other types of UL, those carrying MED12 mutations undergo regression after menopause thus restricting symptomatic UL almost exclusively to women in their reproductive ages. This may point to so far unknown hormonally-regulated factors interacting with the mutated MED12. Hormone-dependent growth also characterizes fibroadenomas of the breast, one of the two other tumors frequently displaying the same type of MED12 mutations, while this is not the case for CLL. However, the unique pattern of MED12 mutations as well as their high prevalence among frequent human tumors prompted us to speculate about factors that might support the occurrence of this type of genetic alterations in a non-random fashion. What can be noted is a hotspot region within exon 2 of the gene which is affected by most single nucleotid exchanges, deletions, as well as indels.
Deduced from the types and patterns of MED12 mutations in human tumors we have presented evidence supporting the idea that the driver mutations of MED12 do not result from selection of random mutational events but rather can be explained by targeted DNA-strand breaks and their repair, respectively. Then, as the second part of our hypothesis, we have advanced the idea that the interaction of nucleic acid sequences of the human microbiome with the common hotspot of MED12 mutations may constitute the initial event. This involvement of infectious agents would also explain the frequent multiplicity of the corresponding lesions at least in UL. Affected by these drivers, the mutated cells may give rise to clonally independent tumors or, even much more frequently, the mutations may not exert a tumor-driving potential as e.g. in case of frame-shift deletions occurring in no more than single cells thus remaining undetected. In case of “active mutations”, additional factors such as the site of origin, angiogenetic support, and the type of MED12 mutation, may endow the resulting monoclonal lesions with a different growth potential.
To stimulate further discussion a possible interaction of a sequence of Staphylococcus aureus with this hotspot has been considered in more detail as depicted in Figure 7 and Figure 8. As to the initial stage of tumor development, the clearance of R-loops resulting from a hybrid helix between a human target cell and bacterial RNA may simultaneously give rise to multiple mutated cells.
This hypothesis, though able to explain main biological characteristics of UL and fibroadenomas of the breast, has some weaknesses and leads to further questions. One point relates to the transmission of the pathogenetic bacteria. Do they act after sexual transmission and ascending infection, respectively? Since parity is well-documented leading to a decreased leiomyoma burden, the argument for reproductive tract infections is not supported. Similarly, parity is also associated with increased risk of postpartum iatrogenic infections. On the other hand, the presence of the bacteria and their nucleic acids has not necessarily to be associated with sexual i.e. ascending transmission and clinically manifest pelvic infections. Interestingly, Staphylococcus forms part of the normal microbiota of some organs and its circulating nucleic acids may thus derive from bacteria/infections of other sites of the body. As to the transfer of such nucleic acids into the host cell, the internalization of naked bacterial RNA or, alternatively, transfer via bacterial extracellular vesicles or target cells that first internalize the bacteria have to be considered. Of note, Staphylococcus is a facultative intracellular pathogen (Sendi & Proctor, 2009) with various types of host cells having the ability to internalize the bacteria (Rollin et al., 2017). Furthermore, it remains to be investigated which mechanisms can lead to an unfolding of short hairpin transcripts and their hybridization to genomic DNA.
In summary, we feel that the exceptional epidemiology of tumors affected by MED12 mutations as well as the patterns of their mutations warrant unusual explanations helping to decipher the etiology and molecular pathogenesis of some of these highly frequent human tumors.
All data underlying the results are available as part of the article and no additional source data are required.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)