Yam-Puc JC, Zhang L, Zhang Y and Toellner KM. Role of B-cell receptors for B-cell development and antigen-induced differentiation [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):429 (https://doi.org/10.12688/f1000research.13567.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
B-cell development is characterized by a number of tightly regulated selection processes. Signals through the B-cell receptor (BCR) guide and are required for B-cell maturation, survival, and fate decision. Here, we review the role of the BCR during B-cell development, leading to the emergence of B1, marginal zone, and peripheral follicular B cells. Furthermore, we discuss BCR-derived signals on activated B cells that lead to germinal center and plasma cell differentiation.
Keywords
B cell receptor, B cell differentiation, B cell development,
Corresponding authors:
Juan Carlos Yam-Puc, Kai-Michael Toellner
Competing interests:
No competing interests were disclosed.
Grant information:
This work was supported by grants from the BBSRC UK BB/M025292/1 and a postdoctoral fellowship from the National Council of Science and Technology Mexico (CONACYT-Mexico) to JCY-P (CVU 49896).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
B cells, the antibody-producing cells, have a protagonist role in the immune response. Although the presence and relevance of antibodies were established more than 100 years ago1 and antibody-producing cells were identified in the mid-20th century2, it took until 1965 for the distinctive B-cell lineage to be recognized3,4. Today, there are still many questions about B-cell differentiation during development and after activation and about the signals that govern such differentiation.
B cells undergo a diversification process during their development in bone marrow and fetal liver, and, as part of this differentiation, B cells rearrange the immunoglobulin (Ig) heavy (H) and light (L) chain gene loci to create a complete Ig molecule5,6. The commitment of the common lymphoid progenitor to the B-cell lineage can be recognized by the expression of the B220 isoform of CD456–8. B cells then develop through several well-characterized stages, ending with the expression of surface IgM and IgD class Ig molecules, which, in association with Igα and Igβ, form the B-cell receptor (BCR) for the antigen5,6. BCR signaling is required for B-cell maturation and survival, and BCR must provide tonic signals, either spontaneously or on interaction with ligands in the environment9–12. After this, B cells emerge to recirculate through secondary lymphoid organs such as the spleen and lymph nodes13. Being well positioned in the secondary lymphoid organs, mature naïve B cells are ready to respond to antigens. Recognizing the antigen through the BCR, B cells are activated and differentiate into plasma cells through extra-follicular differentiation14, or they become germinal center (GC) precursor cells to start GC reactions15,16. The precise signaling mechanisms of B-cell fate decision during this stage are not entirely understood.
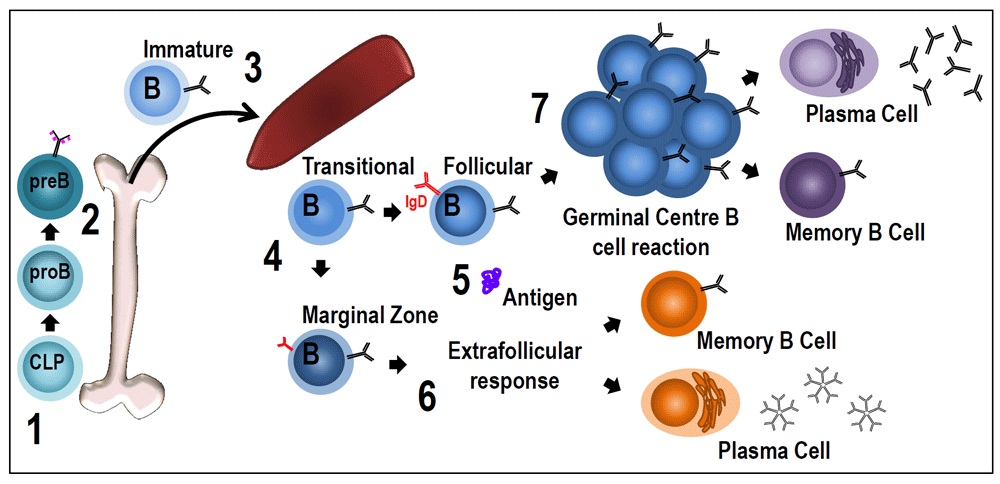
In this review, we will focus on B-cell subsets in the spleen, key steps of differentiation after activation, and, in particular, recent findings about the role of the BCR driving these distinct differentiation stages (Figure 1).
Figure 1. B-cell receptor signaling during B-cell development and for B-cell differentiation after the encounter with the antigen.
(1) Common lymphoid progenitor (CLP) cells commit to the B-cell lineage when they start expressing the B220 isoform of CD45. (2) Pro-B cells undergo DJ rearrangements and become pre-B cells when they express membrane forms of μ heavy chains with surrogate light chains (pink dotted lines) in the pre-B receptor. The transition between pro-B and pre-B cells has been described as being dependent on membrane association of Igα/Igβ complexes and their ability to generate basal signals. It seems that pre-B receptors do not have a ligand-recognition function40,41. (3) After VDJ recombination in the pre-B cell stage, immature B cells pair light chains with μ chains to form monomeric IgM, which is expressed at the cell surface in association with Igα/Igβ to form the B-cell receptor (BCR). Newly formed immature B cells exit the bone marrow to reach the spleen, where, as transitional B cells, they will complete their maturation before entering the follicles or the marginal zone (MZ). (4) BCR signaling strength appears to have a critical role during follicular (Fo) or MZ B-cell differentiation. There is evidence of at least two possibilities29,30,36,42–44: increased BCR signaling could drive differentiation to Fo B cells or to MZ B cells. This could depend on other factors such as the microenvironment or the timing of BCR signaling. (5) Mature naïve B cells are ready to respond to antigens. By specifically binding antigen through the BCR, B cells are activated and differentiate into plasma cells through extra-follicular differentiation (6) or start the T-cell-dependent germinal center (GC) reaction (7). The mechanism of activated B cells entering the GC reaction or undergoing rapid plasma cell differentiation in extra-follicular proliferative foci is controlled by the nature of the interaction between the BCR and antigen. Responding clones that undergo a strong initial interaction with antigen can efficiently differentiate into extra-follicular plasma cells45. However, it has also been shown that B cells expressing higher-affinity BCRs are more competitive to become pre-GC B cells during T–B interaction46,47. This seems contradictory, but timing may be a very important factor. B cells in the GC reaction are selected on the basis of their interaction with the antigen through BCRs. Based on how efficiently B cells present antigen to Fo T helper cells, they are allowed to further differentiate inside the GC. Whether this BCR–antigen interaction results in significant signaling and has a role for selection is not clear.
Brief overview of B-cell receptor signaling
BCR signaling has been intensely studied over the last 30 years, and details of this complex process are still not fully understood. In-depth reviews have been produced by others17–20. In brief, BCR is activated by binding of the antigen. This leads to phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) by the first kinase in the BCR signaling pathway, primarily LYN (part of the src-kinase family). After this, SYK is recruited through its SH2 domain to the phosphorylated Igα–Igβ heterodimer. The higher propensity of ligand-bound BCR molecules to aggregate can enhance their association with src-family PTKs17,20. Once SYK is activated, the BCR signal is propagated via a group of proteins associated with the adaptor protein B-cell linker (BLNK, SLP-65). Phospho-BLNK serves as a scaffold for the assembly of the other components, including Bruton’s tyrosine kinase (BTK), VAV 1, and phospholipase C-gamma 2 (PLCγ2)19. The initiation of the BCR signal is indirectly regulated by at least two non-receptor-associated molecules: B220 and C-terminal src tyrosine kinase (CSK)17.
Additionally, following BCR ligation, tyrosines of the cytoplasmic tail of CD19 are phosphorylated by LYN to create binding sites for the SH2 domains of the p85 adaptor subunit of PI-3K as well as other SH2 domain-containing effectors. Activated PLCγ2 cleaves membrane-associated phosphoinositide PI(4,5)P2 into the second messengers I(1,4,5)P3 and DAG. I(1,4,5)P3 generation causes the mobilization of Ca2+ from intracellular and extracellular stores. Ca2+ signaling is required for the activation of transcription factors such as nuclear factor kappa B (NF-κB) and nuclear factor of activated T cells (N-FAT) by protein kinase C (PKC)21. DAG represents a classic activator of PKC isotypes which regulate the mitogen-activated protein kinase (MAPK) family (extracellular signal-regulated kinase [ERK], c-Jun NH2-terminal kinase [JNK/SAPK], and p38 MAPK); the overall result of these processes drives activation of the B cell, antigen presentation, cytokine production, and cell proliferation and differentiation17–19. In the following, we will discuss the effects of BCR signaling during B-cell development and after the encounter with the antigen.
B1 B cells
Three major populations of mature B cells have been described in mice: B1, marginal zone (MZ), and follicular (Fo) B cells. The phenotypic, microanatomic localization and functional differences that characterize them suggest specialized functions linked to the niches in which they reside5. B1 cells produce polyreactive natural antibodies (NAbs) of relatively low affinity and primarily of the IgM isotype22. NAbs play a critical role in the innate immune response against pathogens, and their absence can lead to increased susceptibility to microbial infections23–25. B1 cells are the major subpopulation in pleural and peritoneal cavities; however, they can also be found in the spleen and other lymphoid organs but at low frequency26. B1 cells consist of two functional specialized subpopulations regarding the expression of CD5: CD5+ B1a and CD5− B1b cells27. However, expression of Blimp-1 also delineates two main coexisting B1 subpopulations in the bone marrow and spleen: B1 Blimp-1hi cells that are dependent on Blimp-1 for maximal natural Ig production and those B1 cells that neither express Blimp-1 nor require it for steady-state antibody production28. Recently, it has been shown that interleukin 17A (IL-17A) promotes B1-cell infiltration into lungs during viral infection, where B1a cells differentiate into IgM-producing plasma cells. This process was promoted through Blimp-1 expression and NF-κB activation25. It is conceivable that the regulation of Blimp-1 expression would also drive the functional role of B1 subsets in sites such as the lung.
What is the role of BCR signaling in B1-cell differentiation? Studies with genetically modified mice indicate that the strength of BCR signaling may control the development or persistence of B cells (or both)29–36. Mutations that enhance BCR signaling strength through the specific deletion of SHP-1 in B cells expand the B1a population. SHP-1 is a protein-tyrosine phosphatase expressed in hematopoietic cells and plays a signal-attenuating role in pathways initiated by many ITAM-containing receptors37–39. The signal-attenuating effects of SHP-1 are mediated primarily via its binding to inhibitory receptors, such as CD5, expressed by B1a cells34. Additionally, enhanced tonic BCR signaling results in an increased B1 B-cell subpopulation and a dysregulated homeostasis of other B-cell subsets33. These findings indicate that BCR signaling is important in fate decisions during B1 cell development, and further studies are needed to better understand these mechanisms.
Marginal zone B cells
MZ B cells contribute about 5–10% of the B cells in the spleen. The “marginal zone” designation refers to the splenic structure that separates the red and the white pulp adjacent to the marginal sinus, where—in both mice and humans—these MZ B cells are in direct contact with blood and its contents5,48. The specialized localization and migration of B cells are strictly regulated under the guidance of different chemokine–chemokine receptor pairs, such as CXCL13–CXCR5, S1PR1, and S1P49–53.
Blood from the primary sinusoids in the spleen perfuses the MZ; the anatomic location of MZ B cells facilitates their role as a rapid first line of defense against blood-borne particulate antigens52,54. After MZ B cells capture the antigen, they transport it to the follicles and deliver it to follicular dendritic cells (FDCs)52,53,55. Furthermore, MZ B cells respond to thymus-independent type 2 antigens producing high quantities of IgM and IgG314,56,57.
Newly formed B cells exiting the bone marrow reach the spleen at a relatively immature stage; these are termed transitional B cells and they need to complete their maturation in the spleen before entering the follicles or the MZ58. It has been described that B cells in the transitional 2 (T2) stage face a decision to mature into either Fo or MZ B cells5,48. However, very recently, it was shown that T1 B cells can differentiate to MZ B cells32. During this differentiation, signaling through the BCR is important for the Taok3-mediated acquisition of membrane expression of ADAM10, which cleaves Notch2 and CD2331,32. MZ B-cell instruction requires triggering of Notch2 on developing B cells by Delta-like 1 (Dll1) expressed by splenic red pulp sinus endothelial cells or MZ reticular cells35,59. How exactly B-cell-positive selection and BCR signaling are causing Taok3 activation and ADAM10 surface expression will require further study32. Furthermore, BCR signaling strength appears to have a critical role during MZ B-cell development; mice lacking secreted IgM displaying increased BCR signaling had increased MZ and decreased Fo B-cell numbers36,42,43. However, some studies reported that MZ B cells need low BCR signaling strength for their differentiation but that transitional B cells with higher BCR signaling strength favor differentiation into Fo B cells29,30,44.
Follicular B cells
Fo B cells are the most prevalent of the three subsets of B cells and the better-studied subpopulation. Their anatomic enrichment in primary follicles gives them their name; however, they are not confined to the follicles and also predominate among the mature populations of B cells in the bone marrow, blood, and other lymphoid organs5. Fo B cells are involved mainly in interactions with T cells, and their responses to T-cell-dependent antigens eventually originate GC reactions16. Many of the mechanisms producing the selection of B-cell subsets, the roles of self- and environmental antigens, and survival signals that drive or maintain them in their proper anatomic and functional niches remain to be elucidated. BCR signaling has been proposed to be crucial in the selection of B1, MZ, and Fo B cells, supported by different genetically manipulated mice where the altered BCR signaling affected different B-cell subsets32–34,36.
Follicular B-cell activation
After Fo B cells encounter the antigen through the BCR, CXCL13–CXCR5 slows down the motility of B cells by promoting membrane ruffling and LFA-1-supported adhesion to facilitate the antigen-recognizing process and enhance B-cell activation60. Meanwhile, surface expression levels of CCR7 on responding B cells increase rapidly to make them more sensitive to CCL19/CCL2151. CCL19/CCL21 is expressed by T-zone reticular cells and extends a gradient to the Fo region. Along the chemokine gradient, antigen-engaged B cells migrate from follicles to T–B border in order to get signals from primed T cells51,61,62. EBI2 drives B cells to move back to the outer follicle and inter-follicular regions63,64. These results indicate interplay between activated BCR downstream signaling and surface chemokine receptors60,61. Although the possibility is less studied on steady state, tonic BCR signaling could also regulate chemokine receptor expression during their anatomic enrichment in specific areas within the secondary lymphoid organs.
Germinal center B cells
After T–B cell interaction, activated B cells either differentiate into plasma cells in the extra-follicular response14 or become GC precursor cells, migrating back into follicles to start GC reactions16. To get co-stimulation from T cells, B cells need to present cognate antigens to T cells in the major histocompatibility complex II (MHC-II) context. B-cell-captured antigen goes through both extracellular and intracellular degradation to become peptides65. These peptides then are assembled with MHC-II molecules and expressed on the B-cell surface as peptide–MHC (pMHC). pMHC–T-cell receptor recognition has an essential role in the process of T–B “pairing” to ensure the specificity of later reactions. Schwickert et al. showed that a higher amount of pMHC help activated B cells, locking T-cell help on the T–B border, thus enabling them to become GC precursor B cells46. CD40L is the most important cognate signal delivered from T cells. In mice and humans, CD40–CD40L ligation is indispensable for the initial formation of GCs66,67 and is needed to maintain ongoing GC reactions68.
BCR affinity also plays an important role during the initial GC B-cell fate decision. The mechanism of activated B cells entering the GC reaction or undergoing rapid plasma cell differentiation in extra-follicular proliferative foci is controlled by the nature of the interaction between the BCR and antigen. Responding clones that undergo a strong initial interaction with antigen can efficiently differentiate into extra-follicular plasma cells and contribute to the rapid early thymus-dependentantibody response45. Although the requirements for GC entry are not stringent69, responding B cells expressing higher-affinity BCRs on their surface are more competitive to become pre-GC B cells during their T–B interaction46,47. High-affinity BCR captures more soluble antigens70 and leads to a higher amount of pMHC expressed on the B-cell surface, which results in a competitive cognate interaction with T cells. The durations of T–B interactions have critical roles in B-cell fate decisions. Recent research shows that ICAM-1/2 adhesion molecules on B cells can secure long-lasting T–B interactions to enhance T-cell help. The expression of ICAM-1/2 could compensate the lack of MHC-II signaling to form GC B cells71.
BCRs of B cells differentiating in GCs have to interact with antigen repetitively. Whether the BCR-antigen interaction in the GC results in significant signaling and what the role of this is are under debate72–74. The interaction is certainly important to test BCR specificity and binding competitiveness, probably mainly in competition with antibodies present in immune complexes on the FDC networks75,76. More important than BCR signaling may be that the affinity of the BCR–antigen interaction will let B cells take up more or less antigen, resulting in more or less efficient positive selection by Fo T helper cells47,74,76.
Much of the knowledge on B-cell differentiation in response to antigen has been gained by using BCR knock-in animals specific for haptens or single epitopes on model proteins46,77. Recent attempts to use complex protein antigens such as influenza hemagglutinin should be better suited to understand the complex competition and crosstalk of many different clones interacting with different epitopes on a natural complex protein antigen78,79. However, for such experiments, it is important to develop methods that will allow the specific analysis of epitope-specific interactions of different B-cell clones. Without an understanding of whether clonal interactions happen because of BCR binding of overlapping epitopes or whether clones develop with less competition because they do bind different epitopes, it is impossible to conclude whether clones with different affinities compete for the antigen80.
Ultimately, it has been estimated that nearly 50% of newly produced auto-reactive B cells can avoid deletion and are induced into an anergic state in peripheral lymphoid tissues81,82. Thus, perhaps B cells in the periphery are not in the same basal state as we thought they should be. The surface expression level of membrane IgM is downregulated on anergic B cells83,84, indicating that their BCR signaling is somehow different from that of normal naïve B cells. Indeed, anergic B cells maintain a distinct gene expression profile81,84 and have been observed preferentially residing in T-zone areas85, indicating that their surface chemokine receptor expression pattern is also different from that of naïve B cells, possibly because of their different signaling. Several studies have found that anergic B cells can be selected to become GC B cells, although these cells previously were thought to be non-responsive84,86. By recruiting anergic B cells into the GC response involving many rounds of mutation, these B cells may mutate away from their original auto-reactivity and become specific for antigens that may have a close relationship with autoantigens86–89. The mechanism—for example, whether anergic and normal B cells will follow the same rules of B-cell selection or which difference an anergic state can bring to B-cell activation and fate decisions at the T–B border—is still not clear.
Concluding remarks
Co-stimulation from T cells plays indispensable roles in B-cell fate decisions after activation and has been studied in some detail. However, BCR signaling strength and patterns not only affect B-cell selection during development but also have been shown to affect the differentiation of B1, MZ, or Fo B-cell populations. Furthermore, BCR signaling strength may affect antigen-induced B-cell activation, migration, and surface co-stimulator molecule expression levels. It seems that there is still work to be done on how differential BCR signaling influences the differential development of B cells and how the various stages of antigen-induced T–B cell interactions affect further B-cell fate decisions.
Competing interests
The authors declare that they have no competing interests.
Grant information
This work was supported by grants from the BBSRC UK BB/M025292/1 and a postdoctoral fellowship from the National Council of Science and Technology Mexico (CONACYT-Mexico) to JCY-P (CVU 49896).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Faculty Opinions recommended
References
1.
Ueber das Zustandekommen der Diphtherie-Immunität und der Tetanus-Immunität bei Thieren.
Dtsch med Wochenschr.
1890; 16(49): 1113–4. Publisher Full Text
2.
Fagraeus A:
The plasma cellular reaction and its relation to the formation of antibodies in vitro.
J Immunol.
1948; 58(1): 1–13. PubMed Abstract
3.
Cooper MD, Peterson RD, Good RA:
Delineation of the Thymic and Bursal Lymphoid Systems in the Chicken.
Nature.
1965; 205: 143–6. PubMed Abstract
| Publisher Full Text
5.
Vale AM, Kearney JF, Nobrega A, et al.:
Development and Function of B Cell Subsets, Molecular Biology of B Cells. in Alt FW, et al Editors. Elsevier.
2015; 99–119. Publisher Full Text
7.
Kondo M, Weissman IL, Akashi K:
Identification of clonogenic common lymphoid progenitors in mouse bone marrow.
Cell.
1997; 91(5): 661–72. PubMed Abstract
| Publisher Full Text
8.
Li YS, Wasserman R, Hayakawa K, et al.:
Identification of the earliest B lineage stage in mouse bone marrow.
Immunity.
1996; 5(6): 527–35. PubMed Abstract
| Publisher Full Text
10.
Lam KP, Kühn R, Rajewsky K:
In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death.
Cell.
1997; 90(6): 1073–83. PubMed Abstract
| Publisher Full Text
13.
Ichii M, Oritani K, Kanakura Y:
Early B lymphocyte development: Similarities and differences in human and mouse.
World J Stem Cells.
2014; 6(4): 421–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
14.
MacLennan IC, Toellner KM, Cunningham AF, et al.:
Extrafollicular antibody responses.
Immunol Rev.
2003; 194: 8–18. PubMed Abstract
| Publisher Full Text
17.
Dal Porto JM, Gauld SB, Merrell KT, et al.:
B cell antigen receptor signaling 101.
Mol Immunol.
2004; 41(6–7): 599–613. PubMed Abstract
| Publisher Full Text
18.
Kurosaki T, Shinohara H, Baba Y:
B cell signaling and fate decision.
Annu Rev Immunol.
2010; 28: 21–55. PubMed Abstract
| Publisher Full Text
20.
Pierce SK, Liu W:
The tipping points in the initiation of B cell signalling: how small changes make big differences.
Nat Rev Immunol.
2010; 10(11): 767–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
21.
Su TT, Guo B, Kawakami Y, et al.:
PKC-beta controls I kappa B kinase lipid raft recruitment and activation in response to BCR signaling.
Nat Immunol.
2002; 3(8): 780–6. PubMed Abstract
| Publisher Full Text
22.
Avrameas S:
Natural autoantibodies: from 'horror autotoxicus' to 'gnothi seauton'.
Immunol Today.
1991; 12(5): 154–9. PubMed Abstract
| Publisher Full Text
23.
Ochsenbein AF, Fehr T, Lutz C, et al.:
Control of early viral and bacterial distribution and disease by natural antibodies.
Science.
1999; 286(5447): 2156–9. PubMed Abstract
| Publisher Full Text
27.
Ghosn EE, Sadate-Ngatchou P, Yang Y, et al.:
Distinct progenitors for B-1 and B-2 cells are present in adult mouse spleen.
Proc Natl Acad Sci U S A.
2011; 108(7): 2879–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
29.
Cariappa A, Takematsu H, Liu H, et al.:
B cell antigen receptor signal strength and peripheral B cell development are regulated by a 9-O-acetyl sialic acid esterase.
J Exp Med.
2009; 206(1): 125–38. PubMed Abstract
| Publisher Full Text
| Free Full Text
34.
Pao LI, Lam KP, Henderson JM, et al.:
B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity.
Immunity.
2007; 27(1): 35–48. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
37.
Matthews RJ, Bowne DB, Flores E, et al.:
Characterization of hematopoietic intracellular protein tyrosine phosphatases: description of a phosphatase containing an SH2 domain and another enriched in proline-, glutamic acid-, serine-, and threonine-rich sequences.
Mol Cell Biol.
1992; 12(5): 2396–405. PubMed Abstract
| Publisher Full Text
| Free Full Text
39.
Yi TL, Cleveland JL, Ihle JN:
Protein tyrosine phosphatase containing SH2 domains: characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12-p13.
Mol Cell Biol.
1992; 12(2): 836–46. PubMed Abstract
| Publisher Full Text
| Free Full Text
40.
Bannish G, Fuentes-Pananá EM, Cambier JC, et al.:
Ligand-independent signaling functions for the B lymphocyte antigen receptor and their role in positive selection during B lymphopoiesis.
J Exp Med.
2001; 194(11): 1583–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
41.
Mårtensson IL, Ceredig R:
Review article: role of the surrogate light chain and the pre-B-cell receptor in mouse B-cell development.
Immunology.
2000; 101(4): 435–41. PubMed Abstract
| Publisher Full Text
| Free Full Text
42.
Waisman A, Kraus M, Seagal J, et al.:
IgG1 B cell receptor signaling is inhibited by CD22 and promotes the development of B cells whose survival is less dependent on Ig alpha/beta.
J Exp Med.
2007; 204(4): 747–58. PubMed Abstract
| Publisher Full Text
| Free Full Text
43.
Horikawa K, Martin SW, Pogue SL, et al.:
Enhancement and suppression of signaling by the conserved tail of IgG memory-type B cell antigen receptors.
J Exp Med.
2007; 204(4): 759–69. PubMed Abstract
| Publisher Full Text
| Free Full Text
44.
Niiro H, Clark EA:
Regulation of B-cell fate by antigen-receptor signals.
Nat Rev Immunol.
2002; 2(12): 945–56. PubMed Abstract
| Publisher Full Text
50.
Förster R, Mattis AE, Kremmer E, et al.:
A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen.
Cell.
1996; 87(6): 1037–47. PubMed Abstract
| Publisher Full Text
54.
Gray D, MacLennan IC, Bazin H, et al.:
Migrant mu+ delta+ and static mu+ delta- B lymphocyte subsets.
Eur J Immunol.
1982; 12(7): 564–9. PubMed Abstract
| Publisher Full Text
55.
Gray D, Kumararatne DS, Lortan J, et al.:
Relation of intra-splenic migration of marginal zone B cells to antigen localization on follicular dendritic cells.
Immunology.
1984; 52(4): 659–69. PubMed Abstract
| Free Full Text
| Faculty Opinions Recommendation
56.
Oliver AM, Martin F, Gartland GL, et al.:
Marginal zone B cells exhibit unique activation, proliferative and immunoglobulin secretory responses.
Eur J Immunol.
1997; 27(9): 2366–74. PubMed Abstract
| Publisher Full Text
57.
Vinuesa CG, Sze DM, Cook MC, et al.:
Recirculating and germinal center B cells differentiate into cells responsive to polysaccharide antigens.
Eur J Immunol.
2003; 33(2): 297–305. PubMed Abstract
| Publisher Full Text
59.
Tan JB, Xu K, Cretegny K, et al.:
Lunatic and manic fringe cooperatively enhance marginal zone B cell precursor competition for delta-like 1 in splenic endothelial niches.
Immunity.
2009; 30(2): 254–63. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
60.
Sáez de Guinoa J, Barrio L, Mellado M, et al.:
CXCL13/CXCR5 signaling enhances BCR-triggered B-cell activation by shaping cell dynamics.
Blood.
2011; 118(6): 1560–9. PubMed Abstract
| Publisher Full Text
61.
Calpe E, Codony C, Baptista MJ, et al.:
ZAP-70 enhances migration of malignant B lymphocytes toward CCL21 by inducing CCR7 expression via IgM-ERK1/2 activation.
Blood.
2011; 118(16): 4401–10. PubMed Abstract
| Publisher Full Text
62.
Okada T, Cyster JG:
B cell migration and interactions in the early phase of antibody responses.
Curr Opin Immunol.
2006; 18(3): 278–85. PubMed Abstract
| Publisher Full Text
63.
Gatto D, Paus D, Basten A, et al.:
Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses.
Immunity.
2009; 31(2): 259–69. PubMed Abstract
| Publisher Full Text
65.
Yuseff MI, Pierobon P, Reversat A, et al.:
How B cells capture, process and present antigens: a crucial role for cell polarity.
Nat Rev Immunol.
2013; 13(7): 475–86. PubMed Abstract
| Publisher Full Text
66.
Allen RC, Armitage RJ, Conley ME, et al.:
CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome.
Science.
1993; 259(5097): 990–3. PubMed Abstract
| Publisher Full Text
67.
Ferrari S, Giliani S, Insalaco A, et al.:
Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM.
Proc Natl Acad Sci U S A.
2001; 98(22): 12614–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
68.
Han S, Hathcock K, Zheng B, et al.:
Cellular interaction in germinal centers. Roles of CD40 ligand and B7-2 in established germinal centers.
J Immunol.
1995; 155(2): 556–67. PubMed Abstract
69.
Dal Porto JM, Haberman AM, Kelsoe G, et al.:
Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced.
J Exp Med.
2002; 195(9): 1215–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
70.
Batista FD, Neuberger MS:
B cells extract and present immobilized antigen: implications for affinity discrimination.
EMBO J.
2000; 19(4): 513–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
77.
Phan TG, Amesbury M, Gardam S, et al.:
B cell receptor-independent stimuli trigger immunoglobulin (Ig) class switch recombination and production of IgG autoantibodies by anergic self-reactive B cells.
J Exp Med.
2003; 197(7): 845–60. PubMed Abstract
| Publisher Full Text
| Free Full Text
80.
Toellner KM, Sze DM, Zhang Y:
What Are the Primary Limitations in B-Cell Affinity Maturation, and How Much Affinity Maturation Can We Drive with Vaccination? A Role for Antibody Feedback.
Cold Spring Harb Perspect Biol.
2017; pii: a028795. PubMed Abstract
| Publisher Full Text
82.
Grandien A, Fucs R, Nobrega A, et al.:
Negative selection of multireactive B cell clones in normal adult mice.
Eur J Immunol.
1994; 24(6): 1345–52. PubMed Abstract
| Publisher Full Text
83.
Quách TD, Manjarrez-Orduño N, Adlowitz DG, et al.:
Anergic responses characterize a large fraction of human autoreactive naive B cells expressing low levels of surface IgM.
J Immunol.
2011; 186(8): 4640–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
85.
Cyster JG, Hartley SB, Goodnow CC:
Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire.
Nature.
1994; 371(6496): 389–95. PubMed Abstract
| Publisher Full Text
86.
Cappione A 3rd, Anolik JH, Pugh-Bernard A, et al.:
Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus.
J Clin Invest.
2005; 115(11): 3205–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
This work was supported by grants from the BBSRC UK BB/M025292/1 and a postdoctoral fellowship from the National Council of Science and Technology Mexico (CONACYT-Mexico) to JCY-P (CVU 49896).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Yam-Puc JC, Zhang L, Zhang Y and Toellner KM. Role of B-cell receptors for B-cell development and antigen-induced differentiation [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):429 (https://doi.org/10.12688/f1000research.13567.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Liu W. Reviewer Report For: Role of B-cell receptors for B-cell development and antigen-induced differentiation [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):429 (https://doi.org/10.5256/f1000research.14737.r32583)
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Liu W. Reviewer Report For: Role of B-cell receptors for B-cell development and antigen-induced differentiation [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):429 (https://doi.org/10.5256/f1000research.14737.r32583)
Tolar P. Reviewer Report For: Role of B-cell receptors for B-cell development and antigen-induced differentiation [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):429 (https://doi.org/10.5256/f1000research.14737.r32582)
NOTE: it is important to ensure the information in square brackets after the title is included in this citation.
Reviewer Report06 Apr 2018
Pavel Tolar,
Immune Receptor Activation Laboratory, The Francis Crick Institute, London, UK; Division of Immunology and Inflammation, Imperial College London, London, UK
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Tolar P. Reviewer Report For: Role of B-cell receptors for B-cell development and antigen-induced differentiation [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):429 (https://doi.org/10.5256/f1000research.14737.r32582)
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)