Keywords
immunoglobulin, CSR, AID, DNA repair
immunoglobulin, CSR, AID, DNA repair
Mammalian adaptive immune responses require B cells to produce immunoglobulins (Igs), commonly known as antibodies, that can recognize a seemingly infinite number of antigens on foreign pathogens. Composed of two heavy (IgH) and two light (IgL) chains that are linked by disulfide bonds, each Ig contains an antigen-binding domain formed from the amino-terminal variable regions of IgH and IgL. The carboxyl-terminal constant (C) region of the IgH chain determines the Ig effector function. Three distinct genomic alterations in the IgH and IgL loci enable B cells to generate the diverse repertoire of Igs: V(D)J recombination, class switch recombination (CSR), and somatic hypermutation (SHM). During V(D)J recombination, developing B cells in the fetal liver and the adult bone marrow assemble the variable coding regions of IgH from variable (V), diversity (D), and joining (J) coding segments. IgL coding regions are assembled from V and J coding segments in either the Igκ or Igλ locus. RAG1/RAG2 endonucleases are required for V(D)J recombination, which forms the primary Ig repertoire and promotes the development of mature IgM/IgD-expressing B cells1,2. Mature B cells with membrane-bound IgM or IgD (B-cell receptor [BCR]) (or both) will migrate to secondary lymphoid organs, such as the spleen, lymph nodes, and Peyer’s patches, where binding of the IgM or IgD to its cognate antigen in the presence of helper T cells will promote CSR and SHM.
CSR reorganizes the IgH gene locus to delete the default Cμ/Cδ constant coding exons for an alternative set of downstream constant coding exons (Cγ, Cε, or Cα)3. The B cell thus will switch from expressing IgM or IgD to IgG, IgE, or IgA. Each Ig isotype regulates different effector functions that are necessary for an effective adaptive immune response4. At the molecular level, CSR is a deletional-recombination reaction that occurs at repetitive DNA regions called switch (S) regions, which precede each constant coding exon except Cδ. The intronic region preceding Cδ is a non-canonical, S-like sequence known as σδ. The expression of Cδ, and consequently IgD, is primarily independent of CSR and results from alternative splicing of a primary transcript that includes Cμ and Cδ; however, recent work has shown that CSR to IgD is a rare event confined to mucosa-associated lymphoid tissues and depends on p53 binding protein 1 (53BP1) and myeloid differentiation primary response gene 88 (MyD88)5.
To initiate CSR, DNA double-strand breaks (DSBs) are generated in an upstream donor S region (for example, Sμ) and a downstream acceptor S region (for example, Sα) (Figure 1). The DSBs are ligated by proteins of the classical-non-homologous end-joining (C-NHEJ) and alternative-NHEJ (A-EJ) pathways, and the sequence between the recombining S regions is excised as an extrachromosomal, circular DNA, which is lost during cell division and DNA replication. Unlike CSR, SHM introduces untemplated point mutations, and occasional deletions and insertions, into the recombined V, D, and J coding exons of IgH and IgL genes at a very high rate (10−2–10−3 base pairs per generation)3,6. These mutations, which occur primarily in complementarity-determining regions, allow the generation of Igs with an increased affinity toward their cognate antigen.

The figure depicts CSR between Sμ and Sα in the immunoglobulin heavy chain (IgH) locus. Activation-induced cytidine deaminase (AID) converts cytidines into uridines in S-region DNA. The dU:dG mismatch is converted into DNA double-strand breaks by either the base excision repair (BER) or the mismatch repair (MMR) pathway. In the BER pathway, uracil DNA glycosylase (UNG) removes the uracil base from the DNA to generate an abasic site, which is recognized and cleaved by the apurinic/apyrimidinic endonuclease 1 (APE1). During MMR, the dU:dG mismatch is recognized by mutS homologue 2 and mutS homologue 6 (MSH2 and MSH6), which recruit the complex of exonuclease 1 (EXO1), mutL homologue 1 (MLH1), and post-mitotic segregation 2 (PMS2) to excise a short patch of DNA that includes the dU:dG mismatch. The DNA breaks are ligated by classical or alternative non-homologous end-joining pathways to generate a recombined Igh locus and an excision circle. Rev1 and 14-3-3 are scaffolding proteins, which are necessary for the assembly of the protein complexes participating in CSR.
Both CSR and SHM require activation-induced cytidine deaminase (AID), a 24-kDa protein expressed primarily in activated B cells7,8. AID, a single-stranded DNA (ssDNA) cytidine deaminase, initiates CSR and SHM by converting deoxycytidine (dC) to deoxyuridine (dU) in recombining S regions during CSR or recombined V(D)J coding exons during SHM. The AID-generated dU:dG mismatch activates DNA repair pathways, including the base excision repair (BER) and mismatch repair (MMR) pathways, which induce DSBs to drive CSR (Figure 1) or error-prone repair to promote SHM9.
This review describes the general mechanisms of CSR and highlights recent data on the localization of AID to S regions and the DNA repair pathways that resolve AID-generated dU:dG lesions. For an overview of SHM, readers are referred to other reviews3,4.
Although S regions and Ig variable coding segments are physiological targets of AID during CSR and SHM, respectively, AID can generate DSBs and mutations in non-Ig genes, such as Myc and Bcl610–13. Despite the markedly lower rate of DSB formation and mutation at these non-Ig genes13,14, the resulting DNA translocations or mutations in these off-target genes contribute to the development of mature B cell lymphomas15–17. Thus, mechanisms target AID specifically to the Ig loci to promote CSR and SHM while restricting AID access to the remainder of the B cell genome to limit off-target DSBs and mutations to maintain genome integrity.
Ig heavy chain constant (CH) exons are organized as independent transcriptional units composed of a cytokine-inducible promoter upstream of a non-coding “I-exon”, the intronic S region, and the corresponding CH exons18. T cell–dependent (for example, cytokines and CD40L) or T cell–independent (for example, lipopolysaccharide) stimuli (or both) activate transcription of recombining S regions (Figure 1), which is absolutely required for CSR. The primary germline transcript is spliced into a mature, polyadenylated transcript with no known protein product and is frequently referred to as a “sterile” germline transcript19. Genetic deletion of specific I-exons abolishes germline transcription and CSR to the corresponding isotype20,21. Germline transcription initiating from the I-exons and proceeding through the S regions to the CH exons creates the ssDNA substrates for AID within the transcribed S regions. Each S region varies in length (1–10 kb) and consists of tandem repetitive units that contain a G-rich non-template strand. Deleting the repetitive units within the S regions or replacing the S regions with small core S-region sequences significantly impairs CSR and demonstrates an essential role for these sequences during CSR22–25. Recent data suggest that the repetitive, G-rich non-template strand forms G-quadruplex (G4) structures that facilitate cooperative AID oligomerization at S regions26. In addition, the tandem repeats of 5′-AGCT-3′ within the core S regions recruit AID and its kinase, protein kinase A (PKA), to the S regions via the 14-3-3 adaptor proteins, which specifically recognize the 5′-AGCT-3′ repeats (Xu et al., 201027; reviewed in Xu et al., 201228).
Germline transcription of S regions creates R-loops, wherein the newly transcribed RNA hybridizes to the template DNA to form a stable RNA:DNA hybrid that exposes the non-template DNA as ssDNA, which is the substrate for AID24,29–32. Inversion of the mouse Sγ1 sequence, which converts the G-rich non-template strand to a G-rich template strand, impairs R-loop formation and CSR without affecting germline transcription24. These data demonstrate the inherent ability of G-rich S regions to form R-loops that likely contain G4 structures, which facilitate AID recruitment26,33.
Although R-loop formation at non-Ig loci may redirect AID activity to other regions of the B cell genome34, AID is significantly enriched at the Igh locus, suggesting that factors beyond R-loop formation also restrict AID to the Igh locus during CSR14,35. AID interacts with RNA polymerase II and its associated proteins, such as Spt536, PAF137, and the FACT histone chaperone complex38. In addition, RNA polymerase II, which has stalled at the repetitive G/C-rich S regions, can recruit the RNA exosome to degrade the nascent RNA transcript and facilitate AID deamination of the non-template and template DNA strands39,40.
Germline transcription is necessary but not sufficient for CSR. In mice that lack the Iγ1 exon splice donor site, CSR to IgG1 was abolished despite active S-region transcription41–43, suggesting that either the RNA processing machinery (for example, spliceosome) or the processed transcripts are required for CSR. CTNNBL1, a component of the spliceosome, interacts with AID and is required for CSR and SHM44. Knockdown of the splicing regulator PTBP2 reduces AID at S regions and impairs CSR45,46. These data demonstrate that the spliceosome plays an essential role in localizing AID to S regions.
Sα RNA expression from a plasmid in trans enhanced CSR to IgA in the B-cell line Bcl1B147, suggesting that spliced, intronic S-region RNA derived from germline transcripts have a functional role during CSR. More recently, these S-region RNAs were shown to recruit AID to S-region DNA sequences33,48. Intronic switch RNAs were known to be spliced from primary transcripts to form lariats that undergo hydrolytic degradation, which is catalyzed by the debranching enzyme DBR149. Knockdown of DBR1 in CH12F3 cells reduces CSR; however, expression of switch RNAs in trans bypasses the lariat debranching step in DBR1 knockdown cells to rescue both CSR and AID recruitment to S regions in a sequence-specific manner33,50. In addition, AID bound directly and selectively to sense S-region transcripts, which were shown to form highly stable four-stranded G4 structures33,50. A putative G4 RNA binding motif in AID was identified and mutations in this domain abrogated AID interactions with G4 switch RNA and consequently the localization of AID to S regions and wild-type levels of CSR33,50. Interestingly, a mutation in the RNA binding motif of AID (G133V) has been identified in patients with Hyper-IgM Syndrome who show severe CSR defects51. From these studies, a new regulatory model for AID localization to S regions was proposed whereby the non-coding, intronic S-region RNA, which is produced following germline transcription and splicing, binds to AID to target AID to sites of DNA recombination (S regions) and promote AID-mediated DNA deamination and CSR in a sequence-specific manner. This model connects the data demonstrating the role of germline transcription and splicing in CSR with the binding of AID to S-region RNA and DNA and identifies a critical function for S-region RNA in CSR beyond germline transcription and splicing (Figure 2).

Upon B-cell activation, germline transcription is initiated from a cytokine-inducible promoter (P) and primary germline transcripts are generated from the I-S-CH sequences, which encode the I-exon, switch (S) region, and constant coding exons (CH). These transcripts are spliced to form a mature non-coding, germline transcript and an intronic S lariat. The latter is further processed by the debranching enzyme DBR1 to form a linear S-region transcript. Linear S transcripts fold into G-quadruplex RNA, which is bound by AID. The complex of S-RNA and AID is guided to transcribed S-region DNA as a result of the complimentary between the S-RNA and the transcribed S region. AID, activation-induced cytidine daminase; DBR1, debranching RNA lariats 1.
Epigenetic modifications of the Igh locus during CSR have been proposed to control the recruitment of AID to S regions (comprehensively reviewed in 52). Changes in histones H4 and H3 methylation patterns have been associated with altered levels of CSR, although the functional significance of these modifications during CSR remains unclear. Conditional deletion of two methyltransferases (Suv4-20h1 and Suv4-20h2), which are responsible for histone H4 lysine 20 di- and tri-methylation (H4K20me2 and H4K20me3), in B cells leads to a 50% reduction in CSR53. AID interacts with SUV4-20H1 and SUV4-20H2 in 293F cells and localizes these methyltransferases to S regions to promote SUV4-20-mediated histone trimethylation in B cells undergoing CSR54, suggesting cooperative targeting of methyltransferases and AID to recombining S regions. H3K9me3 tethers AID to Sμ through its interaction with KRAB domain-associated protein 1 (KAP1) and heterochromatin protein 1 (HP1)55, and combinatorial phospho-Ser10 and acetyl-Lys9 modification of H3 (H3K9acS10ph) mediates AID recruitment to S regions by stabilizing S-region DNA binding of 14-3-3, which in turn interacts with AID56. Enrichment of H3K9me3, histone H3 lysine 9 acetylation (H3K9ac), and histone H3 lysine 4 trimethylation (H3K4me3) at recombining, transcribed S regions57–59 and a reduction in CSR in B cells deficient in Pax interaction with transcription-activation domain protein-1 (PTIP), which is responsible for H3K4 methylation, suggest additional epigenetic mechanisms of regulating AID localization to S regions57. However, the functional relevance of H4K20 and H3K9 methylation in the recruitment of AID to S regions during CSR remains unclear, as some data demonstrate that H3K9 tri-methylation and H4K20 methylation (mono-, di-, and tri-methylation) are reduced at recombining S regions55,60. Additional work is required to decipher the epigenetic code at S regions, which will further elucidate the role of post-translational modification of histones in the localization and stabilization of AID at S regions during CSR.
Although AID localization to and deamination of S-region DNA is required for CSR, additional factors downstream of germline transcription and AID recruitment are necessary for wild-type levels of CSR3,45,61,62. The conversion of deaminated DNA into DSBs requires many proteins from DNA repair pathways that have evolved to respond to general DNA damage. The mechanism by which these factors convert deaminated DNA into recombinogenic DNA repair (that is, CSR) rather than canonical DNA repair (that is, restoration of the dC:dG base pair at the site of deamination) remains unknown. Below, we discuss our current knowledge of the DNA repair pathways that are required for CSR and highlight the role that AID phosphorylation plays in the generation of DSBs during CSR.
CSR requires BER and MMR pathways to generate DNA breaks in recombining S regions. Defects in either BER or MMR alone significantly impair CSR, whereas combined BER and MMR deficiency (for example, UNG−/−MSH2−/−) completely blocks CSR in vitro and in vivo9,63. In the BER pathway for CSR, AID-generated dU in S-region DNA is removed by uracil DNA glycosylase (UNG) to generate an abasic site, which is cleaved by the apurinic/apyrimidinic endonuclease (APE1) to create a single-strand break (SSB) in the DNA9,64–66 (Figure 1). Adjacent SSBs on complementary DNA strands constitute a DSB, which is an obligate intermediate in CSR. Human and mouse B cells with inactivating mutations in UNG exhibit impaired DSB formation at S regions and a severe block in CSR66–69. Impaired recruitment of UNG to recombining S regions in Rev1-deficient B cells reduces CSR in vitro and in vivo70. Likewise, mice heterozygous for an APE1 null mutation and CH12F3 cells with a homozygous deletion of APE1 have significantly diminished CSR71–73.
The AID-generated dU:dG mismatch can also be processed into SSBs through MMR9. In this pathway, an MSH2-MSH6 heterodimer recognizes the dU:dG mismatch and recruits a complex of MLH1/PMS2/EXO1 to repair the mismatch (Figure 1). PMS2 (PMS1 homolog 2) generates a SSB distal to the mismatch and subsequently exonuclease 1 (EXO1) converts the DNA breaks into ssDNA gaps by excising the segment of the DNA containing the dU in a 5′-to-3′ direction3. EXO1 excision of dU-containing sequences on opposite DNA strands thus would generate DSBs that are required for CSR18,74. Consistent with this proposed role for PMS2 and EXO1 in converting deaminated S regions into DSBs, humans or mice with inactivating mutations in PMS2 or EXO1 have significant impairments in CSR because of defects in DSB formation in S regions75,76. PMS2- and EXO1-mediated excision of dU:dG mismatched DNA creates a DSB with a 5′ overhang that can be resolved into a blunt (or nearly blunt) DSB by DNA polymerases (η and θ), which subsequently is used by proteins of the C-NHEJ and A-EJ pathways to complete CSR74,77,78.
Despite the overwhelming genetic and biochemical data demonstrating the role of BER and MMR in CSR, the mechanism by which BER and MMR are subverted (or coopted) to promote recombinogenic repair of S regions rather than canonical repair remains uncharacterized. Hypothetically, AID may generate a high density of dU:dG mismatches within the S regions that cannot be repaired by canonical BER and MMR pathways. To maintain genomic integrity, BER and MMR are shunted toward recombinogenic repair and thus CSR. AID phosphorylation regulates the balance of canonical and recombinogenic repair that is mediated by BER and MMR downstream of AID-dependent deamination of S regions62.
Phosphorylation of AID at Ser38 (pS38-AID) is critical for CSR as mice harboring a homozygous S38A knock-in mutation (AIDS38A/S38A) have a significant reduction in CSR79,80. S38 lies within a consensus cAMP-dependent PKA phosphorylation site80–82. A hypomorphic PKA-RIα knock-in mutant (RIαB) substantially impairs CSR and blocks phosphorylation of AID at S regions83, indicating that PKA is required for AID phosphorylation at S38. Multiple isoforms of protein kinase C (PKC) can phosphorylate AID at S38 in vitro80; however, the regulation of PKC-mediated AID phosphorylation in vivo remains unknown. Although the mutant AID (S38A) protein retains wild-type levels of deaminase activity in vitro and binding to S-region DNA in vivo, AIDS38A/S38A B cells cannot efficiently generate DSBs at recombining S regions62. These data in conjunction with biochemical data demonstrating the indirect interaction of pS38-AID with APE1 strongly suggest that pS38-AID is required for DSB formation62. Endogenous wild-type AID in UNG−/−MSH2−/− B cells or catalytically inactive AID cannot be phosphorylated at S38 and consequently cannot bind to APE1; however, treating these cells with ionizing irradiation to induce DSBs restores both AID phosphorylation and APE1 binding, suggesting that the conversion of AID-dependent S-region DNA deamination into single-strand breaks by BER (APE1) or MMR (PMS2/EXO1) is required for AID phosphorylation. Thus, AID phosphorylation at S38 is required for, and dependent on, DNA breaks62. These findings suggest the existence of a positive feedback loop wherein a low density of DNA breaks leads to AID phosphorylation, APE1 binding, and additional DNA breaks, which in turn activate more AID phosphorylation (Figure 3). Consistent with this model, ATM, a serine/threonine protein kinase that activates DNA repair pathways in response to DSBs, is required for wild-type levels of AID phosphorylation and APE1 interaction62. This model uncovers a previously undescribed role for ATM as a molecular rheostat that couples targeted DNA double-strand break formation with non-canonical, recombinogenic DNA repair to promote Ig gene diversification (Figure 3).
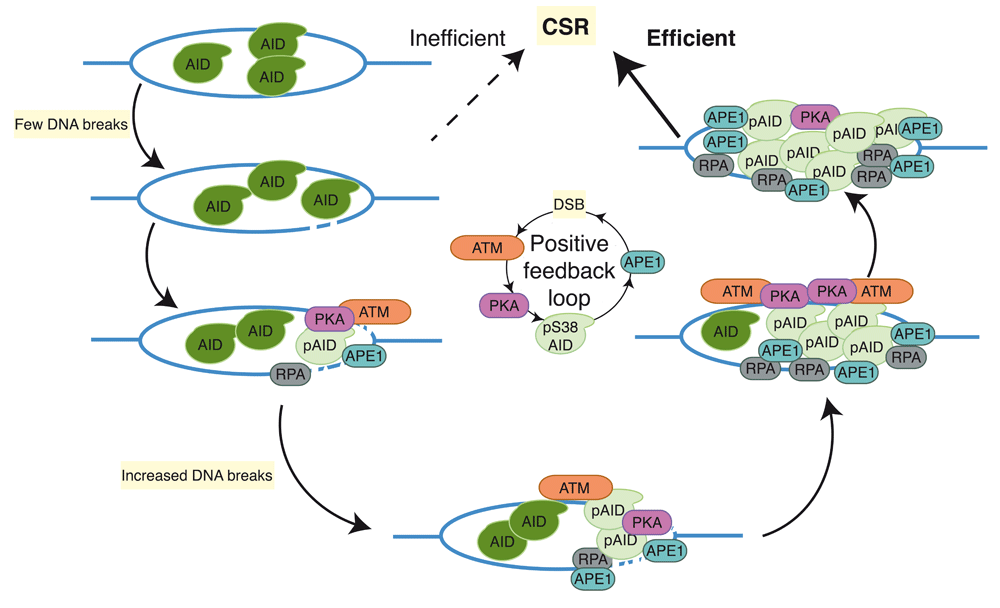
AID-mediated deamination of S regions generates DNA breaks that induce PKA-dependent AID phosphorylation at serine-38 (pS38-AID) and subsequent binding of APE1 and RPA to pS38-AID. Recruitment of APE1 to S regions generates additional DNA breaks, inducing additional AID phosphorylation through an unidentified ATM-dependent mechanism of activating PKA. AID, activation-induced cytidine daminase; APE1, apurinic/apyrimidinic endonuclease 1; ATM, ataxia telangiectasia mutated; CSR, class switch recombination; PKA, protein kinase A; RPA, replication protein A.
CSR requires joining DSBs in donor and acceptor S regions that may be separated by over 100 kb; however, some DSBs within an S region may be joined to another DSB within the same S region, resulting in an internal deletion rather than productive CSR45,84,85. In addition, DSBs in S regions can be ligated to a DSB on another chromosome to generate a chromosomal translocation45. The molecular mechanisms that promote the end joining of DSBs in distal S regions rather than canonical DNA repair, internal deletions, or chromosomal translocations remain largely unknown. During CSR, the Igh locus is re-organized into transcriptionally active loops, wherein I-promoters and regulatory enhancers (Eμ and Eα) are positioned close to one another to promote transcription, accessibility, and synapsis of recombining S regions84–88. Productive CSR is observed in B cells that have S regions replaced by I-SceI restriction sites89, suggesting that the general cellular DNA damage repair pathways, which function in the synapsis and long-range end-joining of S-region DSBs, are required for the resolution of DSBs in S regions and productive CSR89. Following the introduction of DSBs in the S regions, ATM and its substrate 53BP1 are thought to promote S-S region synapsis and recombinogenic repair60,90,91. In the absence of ATM, DSBs at IgH and chromosomal translocations involving IgH are increased and CSR is decreased90–92. Recently, 53BP1 was shown to facilitate S-S synapsis, as Eα interactions with Eμ and the γ1 promoter are reduced in 53BP1−/− B cells that are stimulated with LPS or LPS+IL460.
ATM plays a role not only in the stabilization of S-region DSBs through the proposed synapsis and joining of S regions but also in the generation of DSBs through the phosphorylation of AID and the subsequent interaction of AID with APE162,90–92. Activation of ATM kinase activity requires binding of the Mre11/RAD50/Nbs1 (MRN) complex to DSBs, which induces ATM-dependent phosphorylation of proteins mediating cell cycle checkpoints (for example, p53) and DNA repair, such as H2AX, MDC1, Nbs1, and 53BP160,93,94. The DNA damage response initiated by ATM promotes the assembly of macromolecular foci flanking DSBs and provides docking sites for DNA repair proteins to bind and stabilize DNA ends to promote recombinogenic repair during CSR. Null mutations in ATM substrates impair CSR and increase chromosomal abnormalities and translocations90–92. 53BP1 deficiency leads to the most robust defect in CSR95–97. Mutation or deletion of 53BP1 results in a 90% defect in CSR with a significant proportion of chromosomal aberrations involving the Igh locus92,98 as well as a high frequency of Sμ internal deletions in cells stimulated for CSR99. 53BP1 promotes the synapsis and long-range joining of S regions60 and protects DNA ends from end resection to direct DNA repair toward NHEJ100,101. Consistent with these roles for 53BP1, ATM-mediated phosphorylation of 53BP1 recruits Rap-1 interacting factor (Rif1) to sites of DNA damage to protect DNA ends from resection and to promote DNA repair102. Accordingly, Rif1-deficient B cells are significantly impaired in CSR102 (Figure 4). Additionally, 53BP1 can be recruited to S-region DSBs through ATM-independent pathways. 53BP1 interacts with H4K20me2 at sites of DNA damage. Depletion of the histone methyltransferase MMSET in the CH12F3 B cell line decreases both H4K20me2 levels and 53BP1 accumulation at S regions, thereby impairing CSR103,104. Furthermore, the recruitment of 53BP1 to DSBs has been shown to require the RNF8- and RNF168-dependent histone ubiquitination pathway. RNF8−/− and RNF168−/− B cells have decreased 53BP1 at S regions and a concomitant reduction in CSR105–108. Because CSR is more dramatically reduced in 53BP1-deficient B cells as compared to ATM-, H2AX-, MDC1-, or RNF8-deficient B cells, 53BP1 also has a function during CSR that is independent of the ATM/γH2AX/MDC1/RNF8 DNA damage response90,91,96,97,105–110. Data showing reduced Eα interactions with Eμ and the γ1 promoter in LPS- or LPS+IL4-stimulated 53BP1−/− B cells demonstrate a role for 53BP1 in S-S synapsis during CSR60.

ATM directly or indirectly phosphorylates proteins (for example, H2AX, MDC1, 53BP1, and AID) and stabilizes protein complexes that aid in the formation and resolution of DSBs during class switch recombination. A-EJ, alternative end-joining; AID, activation-induced cytidine daminase; BER, base excision repair; cNHEJ, classical non-homologous end-joining; DSB, double-strand break; HR, homologous recombination; MMR, mismatch repair.
More recently, BRCT-repeat inhibitor of hTert expression (BRIT1) has been implicated as a novel effector of the DNA repair phase of CSR111. BRIT1 is a ubiquitously expressed protein that is rapidly recruited to DSBs after ionizing radiation through its C-terminal BRCT repeat domain, which is necessary for its interaction with phosphorylated H2AX (γH2AX)112. As predicted, successful CSR requires BRIT1 interaction with γH2AX at recombining S regions111. In addition, the BRIT1-γH2AX pathway is further modulated by the interaction of γH2AX with MDC1 in CSR. Although BRIT1 or MDC1 deficiency alone leads to a moderate reduction in CSR, loss of both BRIT1 and MDC1 together markedly impairs CSR111. Thus, BRIT1 likely serves as a scaffold to recruit factors that resolve DSBs at S regions downstream of ATM (Figure 4).
Homologous recombination (HR) and NHEJ are the two major pathways for DSB repair in mammalian cells113. HR is restricted to the S/G2 phase of the cell cycle and requires large stretches of homology, whereas NHEJ is active throughout the cell cycle and requires little or no homology. Since CSR-associated DSBs are observed primarily during the G1 phase of the cell cycle and do not have extended stretches of homology, NHEJ is generally considered the major pathway in the joining of DSBs during CSR95,113. Consistent with this, mutations in the canonical NHEJ components Ku70/Ku80 heterodimer (Ku), XRCC4, and DNA ligase IV (Lig4) severely compromise CSR, while mutations in non-canonical NHEJ proteins such as DNA-dependent protein kinase catalytic subunit (DNA-PKcs), Artemis, and XRCC4-like factor (XLF or Cernunnos) increase chromosomal translocations even though CSR is not severely impaired95.
B cells lacking core NHEJ components are capable of residual CSR, mediated by microhomology-biased A-EJ pathway (Figure 4). Although A-EJ is a poorly defined DNA repair mechanism guided by microhomology between two DSBs, factors from other DNA repair pathways, including XRCC1, Ligase III, Mre11, Parp1, and CtIP, have been shown to be necessary for A-EJ during CSR114–117. More recently, Rad52, an HR repair factor, was shown to facilitate microhomology-mediated A-EJ that favors intra–S region recombination and competes with Ku to mediate inter–S region DSB recombination118. Whether A-EJ is physiologically necessary in order to complete a productive class switch reaction is extensively discussed in 95.
While C-NHEJ and A-EJ are the primary end-joining pathways ligating DSBs within IgH during CSR, HR has been proposed to repair AID-induced off-target DSBs. Deficiency of the Rad51 paralog XRCC2, a key component of HR-mediated repair, significantly enhances AID-dependent genome-wide DNA damage119,120. Notably, AID-expressing human chronic lymphocytic leukemia cells are hypersensitive to HR inhibitors and this is possibly due to AID-dependent synthetic cytotoxicity from unrepaired DSBs at non-Ig loci121. Thus, HR is essential for repairing AID-generated DSBs and dysregulated AID activity may provide a novel therapeutic approach to treat B cell malignancies.
The discovery of AID as a master regulator of CSR and SHM revolutionized our understanding of Ig gene diversification and the mechanisms regulating genome integrity. The physiological targets of AID during CSR and SHM are almost exclusively restricted to S and V regions of the Ig loci, but AID can deaminate in vitro any transcribed substrate and damage in vivo many non-Ig genes, threatening genomic stability in B cells. However, B cells have evolved mechanisms that promote AID-dependent mutagenic and recombinogenic DNA repair within Ig loci while faithfully repairing collateral damage at non-Ig loci using canonical, conserved DNA repair pathways. Unlike V(D)J recombination, CSR has coopted the general DNA damage response to simultaneously generate and resolve DSBs within S regions. BER and MMR are essential, complementary pathways for CSR. ATM functions as a generator of DSBs in S regions, an essential signaling molecule to mobilize DNA repair proteins, and a scaffold for these proteins to resolve DSBs. As additional CSR factors, such as RNF8/168 and BRIT1, are identified, we will further understand the genetic and molecular mechanisms regulating the formation and repair of DSBs during CSR.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)