Keywords
TDG, DNA methylation, human neural progenitors,
TDG, DNA methylation, human neural progenitors,
Coordinated changes to the epigenome are known to be essential for lineage specification and maintenance of cellular identity. DNA methylation and histone modifications critically contribute to epigenetic maintenance of chromatin structures and gene expression programs. DNA methylation can silence genomic regions, directly or indirectly, and play an important role during mammalian development. Loss of methylation in specific locations is associated with differentiation towards specific germ layers as binding of several transcription factors is strongly associated with specific loss of DNA methylation in one germ layer, and in many cases a reciprocal gain in the other layers. However, the mechanism for the lineage related site specific demethylation is not currently known. A major open question is whether this is the result of an active or passive demethylation after repeated cell division1,2. Promoters with low CpG content are more likely to be methylated in human embryonic stem cells (ESCs). Conversely, these same promoters are demethylated and actively expressed during differentiation in a cell-type-specific manner3–5. Demethylation can occur by a passive mechanism in which the normal function of DNMT1/UHRF1 is insufficient or disrupted2,6. Alternatively, evidence for the existence of an active mechanism in which the cytosine modifications are enzymatically removed is accumulating1,7–9. Which of these mechanisms is responsible for demethylation changes in early human development is not currently known.
We and others have shown that standard differentiation protocols of hPSC leads to derivation of an embryonic-like cell rather than a mature, postnatal-like cell10. We previously identified a group of embryonic related genes which are differentially expressed in the PSC progeny of all three lineages and in tissues of the early gestation period rather than in their respective cell types of later developmental stages. Among these genes, we identified Thymine DNA Glycosylase (TDG), a gene that was recently implicated in active DNA demethylation10. Unlike other glycosylases, TDG is essential for embryonic viability as TDG null embryos die around E11.5-12.5 of internal hemorrhage8. The lethal phenotype was also associated with aberrant promoter methylation and imbalanced histone modifications. In addition there is evidence that levels of this enzyme are linked to progression through specific cell cycle stages. Here we provide evidence that that TDG regulates cell cycle related gene expression in human neural progenitors (NPCs) derived from hPSCs and controls their capacity for differentiation towards neurons and glia. These observations suggest that TDG and active demethylation play an important role in hPSC cell cycle regulation and differentiation.
We originally identified TDG in a screen for genes that were consistently differently expressed between human pluripotent derivatives and their in vivo counterparts10. This screen identified a number of genes that were persistently expressed in pluripotent derivatives, and therefore suggestive of an early embryonic state, or genes that failed to be induced in pluripotent derivatives but were highly expressed in tissue derived cells. TDG was expressed significantly higher in neural progenitors generated from pluripotent stem cells as opposed to the same cell type derived from fetal brain (Figure 1A, shown in Log2 scale).
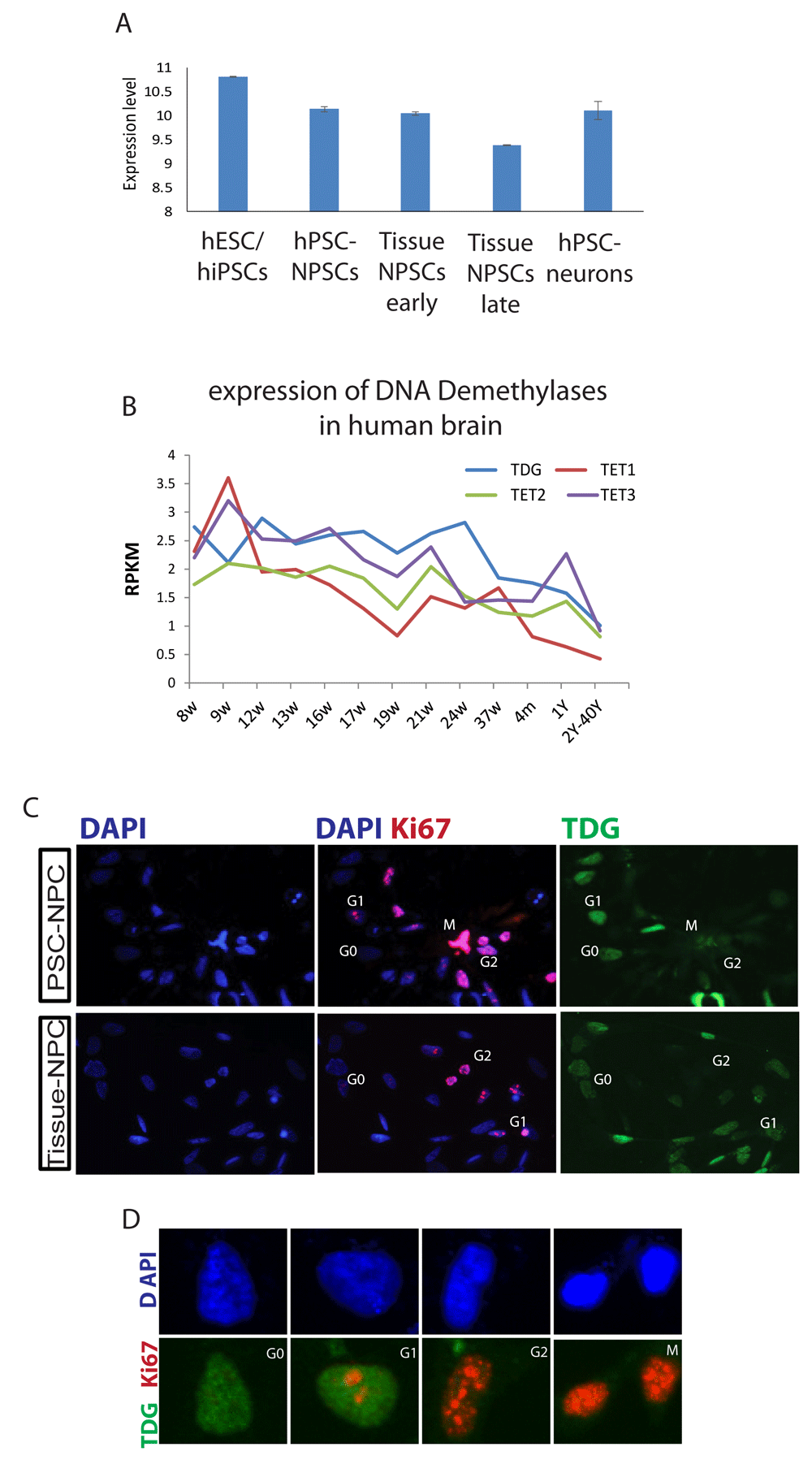
A, Average expression of the TDG microarray probe in hPCS, hPSC derived NPCs and neurons and tissue derived NPCs, adapted from 10. Data presented are the average of at least three independent samples and normalized by log2 according to 13. B, RNA-seq analyses for the expression of TDG, TET1, 2 and 3 adopted from the Allen Institute's Brainspan developmental transcriptome database displayed as log-scale reads per kilobase measured (log2 RPKM) across the developing human brain. C, Immunofluorescent (IF) staining for KI67 and TDG expression in NPCs derived from human pluripotent stem cells (PSC-NPCs) or human brain tissue (Tissue-NPCs). D, Magnified images (from panel C) of representative nuclei for different cell cycle stages. Images were taken at 20X magnification.
The Allen Brain Atlas created by the Allen Institute provides gene expression data from various brain regions across both development and through adulthood. As shown in Figure 1B, TDG is expressed most highly in the brain in utero, and then falls after birth and stays low throughout adulthood. The same was true for the Ten–Eleven Translocation (TET) family of dioxigenases7, suggesting that DNA demethylation is primarily performed in utero. It is also possible that DNA demethylation by TDG and TETs function is linked to proliferation, which is known to decrease at birth relative to that found in utero. Because of this and previous data suggesting TDG could potentially regulate the cell cycle, we stained neural progenitors made from human pluripotent stem cells or derived from tissue for TDG and Ki67. The cell cycle phase for this assay was determined based on a previously reported KI67 staining pattern within the nucleus11. TDG was also previously reported to be tightly regulated during the progression of the cell cycle as its level is rapidly downregulated by ubiquitination in the S phase of the cell cycle in cellular models such as HeLa and fibroblasts and re-expressed in G212. Here, we found a similar result, namely that TDG protein levels appear to correlate with G0/G1stages of the cell cycle (Figure 1C and D).
To investigate the role of TDG in early human development, we used siRNA mediated knock-down (KD) of TDG in neural progenitor cells (NPCs) derived from hPSC (Figure 2A and B). To determine whether TDG KD led to expected changes in 5-carboxylcytosine (5caC) and 5-formylcytosine (5fC) DNA residues, immunostaining for these markers was performed. As expected, silencing TDG led to an increased intensity of 5caC and 5fC DNA, with no change in 5-Hydroxymethylcytosine (5hmC) DNA (Figure 2C).
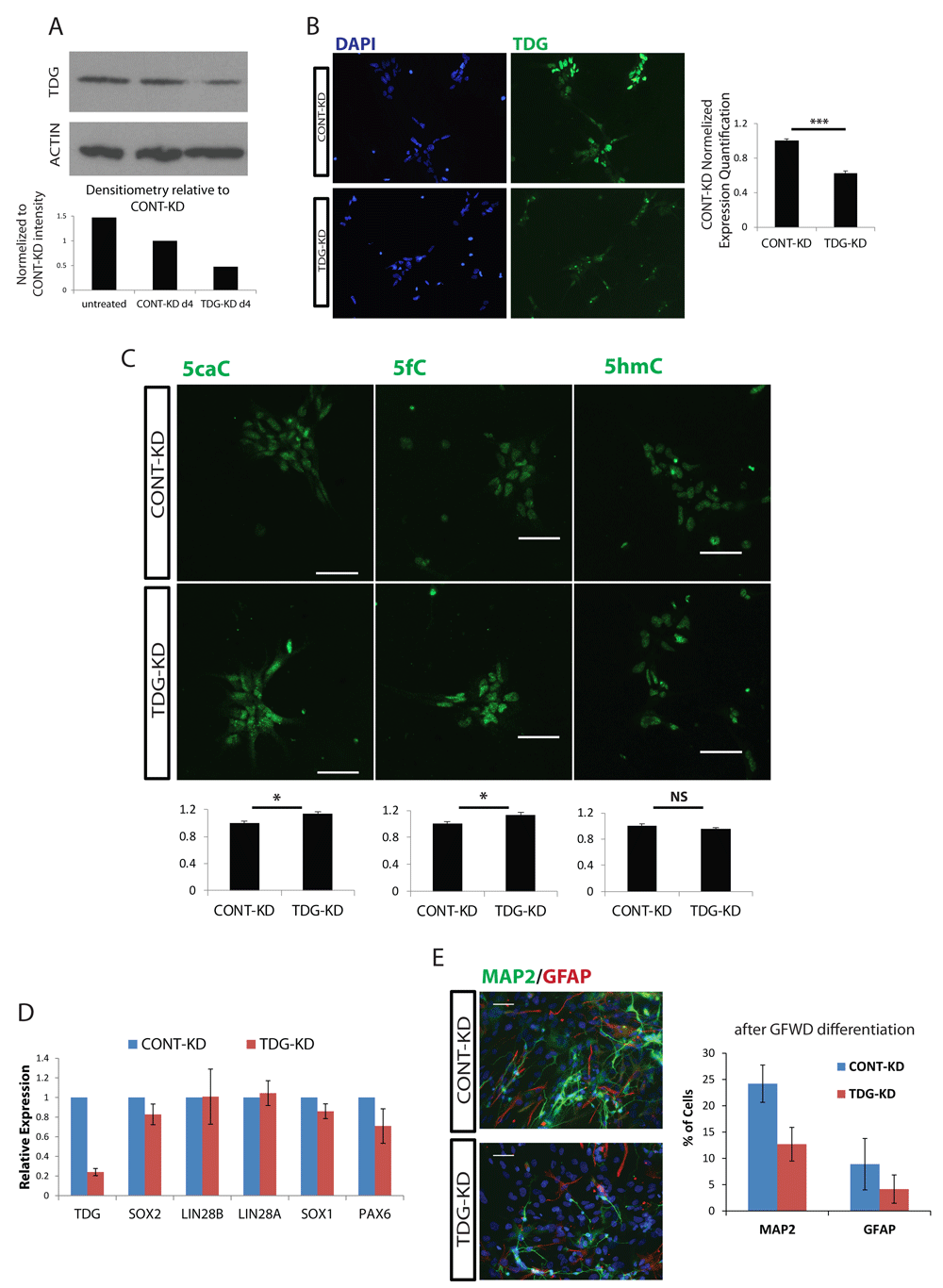
A, TDG protein expression level at day 4 post siRNA transfection, measured by Western blot (top). B, Immunostaining for TDG in NPCs following siRNA transfection, and quantification (right). C, Top: Representative immunofluorescence of DNA methylation modifications. Bars represent 50µm. Bottom: ImageJ quantification of TDG-KD normalized to CONT-KD. Quantification was performed for over 100 nuclei across at least 5 images. Error bars represent standard error of the mean normalized to CONT-KD. D, Expression levels of NPC markers measured by qRT-PCR, normalized against the relative levels of GAPDH and compared to CONT-KD, error bars represent standard error of the mean of 3 knockdown experiments. E, 4 Days post siRNA transfection, NPCs were induced to terminally differentiate by growth factor withdrawal (GFWD). Left: representative IF of 3 Weeks neural differentiation. Efficiency measured by the ratio of percentage of MAP2 (neuron)/GFAP (glia). Bars represent 50µm. Right: quantification of n = 3 from at least three separate knockdown/differentiation experiments. p-values were calculated with Student's t test: * = p<0.05, ns=not significant.
RT-PCR for genes typical of the NPC state showed essentially no change in TDG KD cells (Figure 2D). Though downregulating TDG levels showed no influence on NPC identity, we further tested whether lower TDG levels affects differentiation. Four days post TDG KD treatment, NPCs were induced to further differentiate using the growth factor withdrawal method (removal of self-renewal supporting growth factors EGF, bFGF). Three weeks after induction of differentiation, we analyzed the percentage of MAP2/GFAP positive cells, which represent the differentiation expectancy towards the neuronal/glial lineage respectfully. We found that though the neural/glial ratio remained similar, the total differentiated cell percentage was lower than in control (Figure 2E), indicating a failure to properly differentiate upon silencing of TDG. Typically, such a differentiation block would be due to aberrant differentiation or due to prolonged proliferative stimulus.
We also looked for gene expression changes following TDG-KD in NPCs by RNA-SEQ. 355 genes were differentially expressed by 1.5 fold across 3 independent experiments (Figure 3A). Using the DAVID annotation tool14, we classified those genes into functional groups (Figure 3B). Of the most significantly enriched functional annotations identified, we found 34 cell cycle related genes. Among those, genes which are major players in mitosis, CDK1, CDK10, SKP2 were upregulated. In contrast, other genes like CDC25B and CDKN1C (p57), which are inhibitors of cell cycle progression, were downregulated (Figure 3C).

A, Differential gene expression of n = 3 siRNA knockdown experiments. Scatter plot of the group average FPKM (log2) for all genes mapped above the background cutoff, differentially expressed genes (over 1.5 fold change; p<0.05) are highlighted in red and green. B, Functional annotation of differentially expressed genes shows significant change in genes related to cell cycle, regulation of apoptosis and structural genes. C, Cell Cycle related differentially expressed list of genes and the relative fold change.
To validate that silencing of TDG by siRNA led to changes in DNA demethylation, we performed Methylase-assisted bisulfite conversion PCR (MAB-PCR) to probe for the presence of the 5mC and 5hmC in a gene whose expression changed upon siRNA-mediated knockdown of TDG. MAB-PCR takes advantage of an enzyme and bisulfite-conversion sequencing to identify the relative abundance of 5caC and 5fC nucleotides (Figure 4A). This allows for a measure of TDG activity, as TDG is known to use its glycosylase activity to finish the demethylation process to convert 5caC and 5fC to the fully demethylated state. To determine whether TDG activity can regulate the methylation and gene expression, we looked specifically at a gene whose expression was affected in TDG-KD cells. EGR1, the early growth response gene is known to be dynamically regulated by a variety of mechanisms, including DNA Methylation at an upstream CpG island (Figure 4B)15. We analyzed a segment of a CpG island upstream to the EGR1 transcription start site (TSS site). This locus was chosen as it was reported to have the highest distribution of 5fC, 5caC around the TSS.
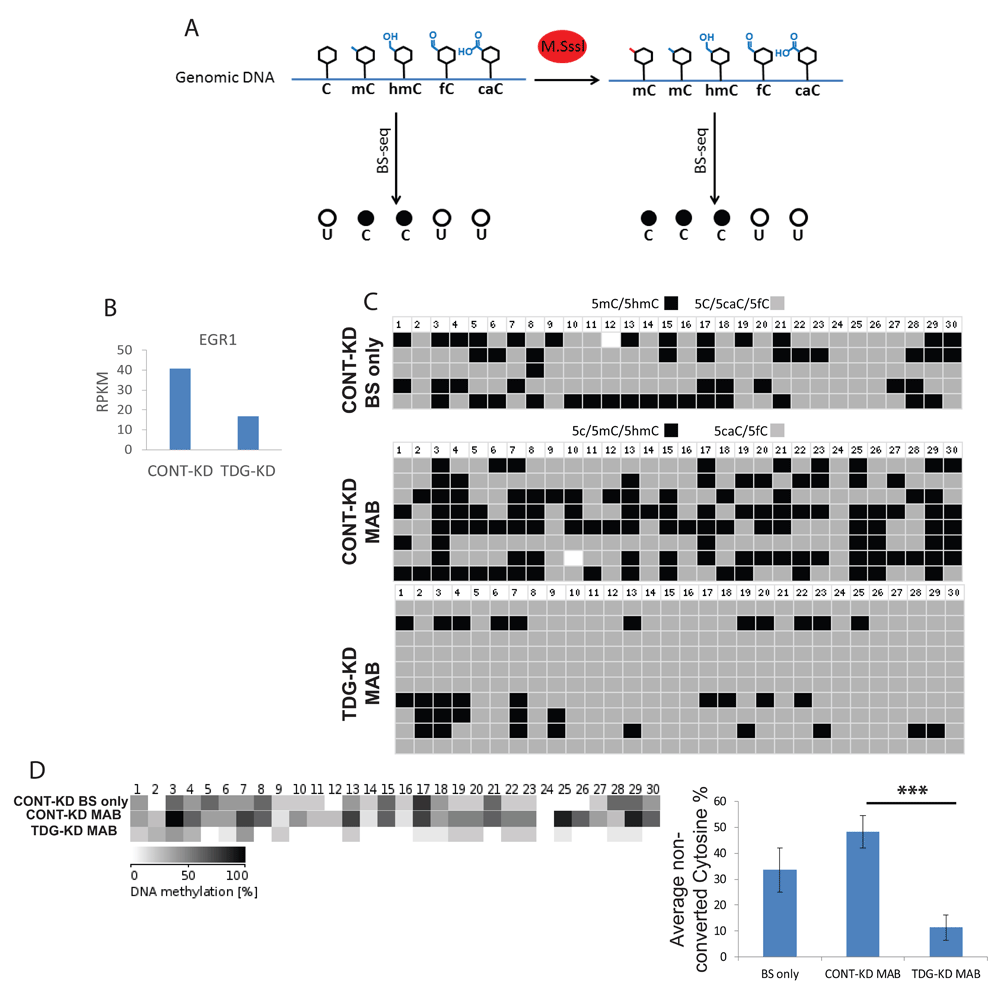
A, Schematic illustration of the sequencing of methylase treated compared to non-treated bisulfite converted transcripts. B, EGR1 expression levels are downregulated following TDG knockdown as measured by RNA-seq (described in Figure 2). C, Sanger sequencing of the CpG island upstream to the EGR1 TSS following either bisulfite conversion (BS only; top) or MAB treatment (bottom two) shows higher abundance of 5cAC, 5fC in TDG deficient cells; numbers indicate CpG dinucleotide position. D, Summary visualization (left) and quantification (right) of the abundance of non-converted residues described in Figure 4C. Error bars represent standard error of the mean methylation level of at least 5 sequenced samples, p values were calculated with Student's t test: *** = p<0.001.
This analysis showed that silencing of TDG by siRNA led to a dramatic accumulation of 5caC and 5fC in a CpG island directly upstream of the start site of EGR1 transcription (Figure 4C and D). This experiment provided evidence that TDG not only regulates DNA demethylation, but also that this can influence gene expression. The proportion of genes differentially regulated by TDG-mediated DNA demethylation remains unclear until a genome-wide analysis can be performed.
We further tested whether TDG-KD in NPCs affects entrance into the cell cycle by Ki67 staining, and found that downregulating TDG resulted in a higher percentage of proliferating cells when this enzyme was knocked down (Figure 5A). Co-staining of Ki67 with TDG showed that TDG is downregulated with cell cycle progression, as higher TDG expression is observed in G0/early G1 cells, and downregulated with cell cycle progression (Figure 2D). We also measured cell cycle dynamics by Flow Cytometry (FACS) upon TDG silencing. This high-throughput method allowed for an accurate determination of the effect of siRNA on TDG, and showed that the proportion of cells in S phase was significantly decreased, while the proportion in G2/M was increased (Figure 5C). Taken together, it seems clear that TDG plays a role in human pluripotent stem cell cycle regulation.
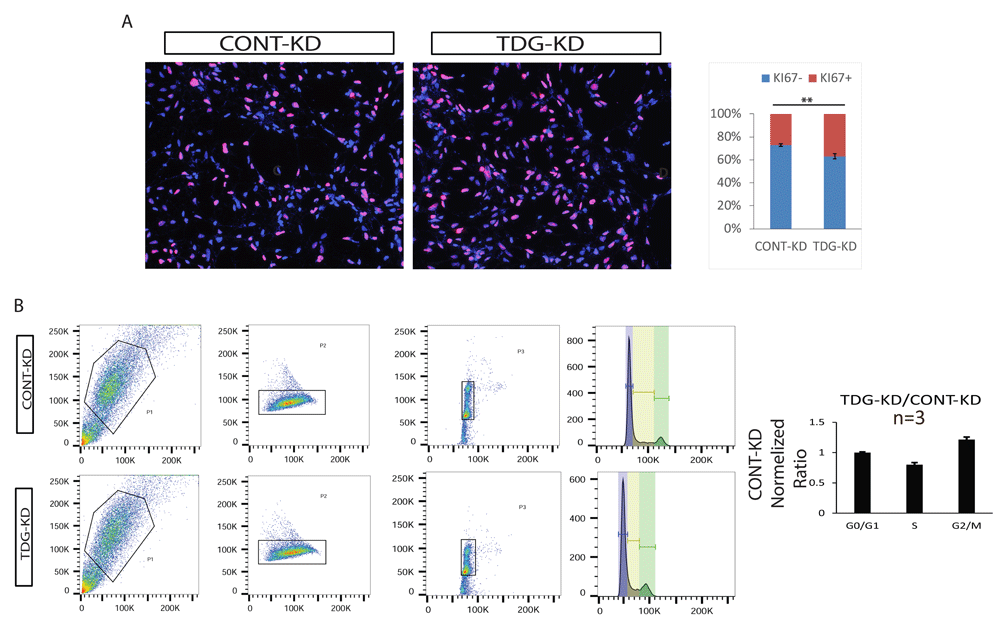
A, TDG downregulation results in higher fraction of cells entering mitotic cell cycle, based on KI67 positive cells staining compared to CONT-KD. Left: representative IF (images were taken at 10X magnification). Right: quantification of over 400 cells from five separate fields. Error bars represent standard error of the mean. p-values were calculated with Student's t test: **=p<0.01. B, TDG downregulation results with a change in cell cycle progression as G2/M increased. Left: representative flow analysis (out of four independent TDG-KD experiments analyzed by flow cytometry) of TDG-KD NPC cell cycle compared to CONT-KD NPC, based on DNA content staining with PI. Right: Quantification of cell cycle phases from 4 separate TDG knockdown experiments
Despite all the analyses above, it was not clear whether silencing of TDG affects the cell cycle through its ability to regulate the terminal step of DNA demethylation. It is formally possible that the DNA glycosylation activity of TDG is used for other substrates besides methylated cytosine, for instance in DNA repair. It is also possible that another domain of TDG regulates cell cycle progression by another unknown mechanism. To attempt to link DNA demethylation by TDG to regulation of the cell cycle, we needed a system that could allow for simultaneous labeling of DNA demethylation intermediates and cell cycle markers. We generated hESCs which expressed the Fluorescence Ubiquitination Cell Cycle Indicator (FUCCI) transgene reporter system by lentiviral transduction16. In this system, cells which are in the G1 stage express Ctd1, which is conjugated to mCherry, while cells in S/G2 expressed Geminin which is conjugated to visible green protein mVENUS. Cells entering the DNA replication stage at the end of G1 express both markers and emit yellow light (Figure 6A). We first verified that the level of TDG was tightly regulated during cell cycle progression in hPSC as in other reported cell systems since hPSC display a unique cell cycle pattern. We found high levels of TDG in early G1 which are downregulated with cell cycle progression (Figure 6B). Interestingly, we found that 5caC and 5fC were both induced in early S phase cells, while 5hmC was reduced (Figure 6B) as was reported before. This indicates that the global state of DNA demethylation is tightly correlated to progression of the cell cycle. Furthermore, all these data on the effect of TDG on cell cycle serve to explain why silencing of TDG led to defective neuronal and glial specification in NPCs (Figure 2D).
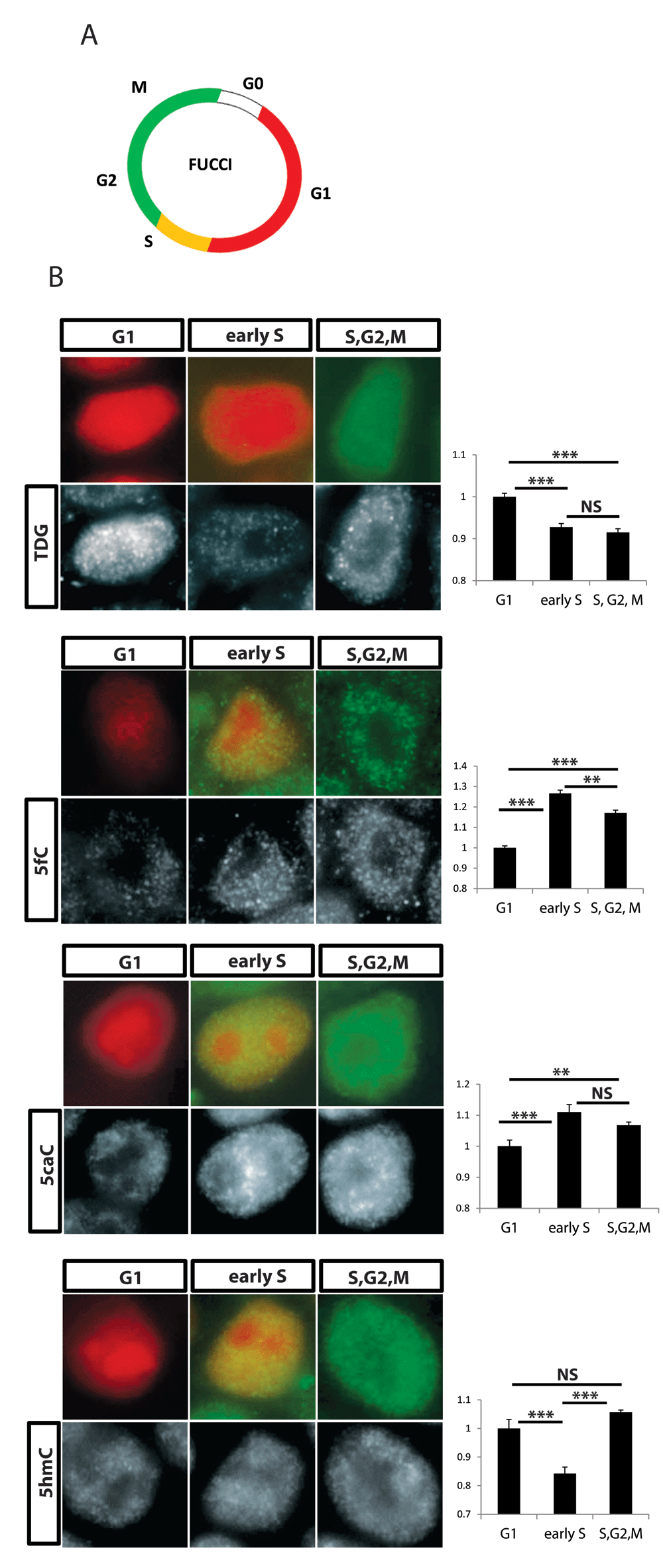
A, Illustration of the FUCCI cell cycle reporter system expression. B, Co-staining for particular cell cycle phase with antibody against either TDG or methylation intermediate modifications. Left: representative nucleotide for each cell cycle stage (Pics were taken using a 20X magnification and a cropped single nucleotide is presented) Right: intensity of staining was quantified using ImageJ for over 500 cells, values normalized to G1 (Y Axis) are presented. p values were calculated with Student's t test: **=p< 0.01, ***=p<0.001, ns=not significant.
The data presented here confirm and extends previous findings that TDG and DNA demethylation can play a role in proper progression through the cell cycle. These results could be particularly relevant for the nervous system, where we provide evidence that TDG and TET mediated demethylation appears to diminish across development. This correlates with both proliferative rate and TDG expression, and could have important consequences to the rate of developmental progression. The big question remaining from this work is how DNA demethylation plays a role in progression of the cell cycle. When DNA is replicated it is thought that the new daughter strand is methylated according to the hemi-methylation pattern on the sister strand by maintenance DNA Methyltransferases (DNMT1). Less clear is what happens to portions of the genome that are hemi-methylated hydroxymethylated nucleotides. The change in proportions of 5caC and 5fC across the cell cycle could indicate that these modified nucleotides are simply erased through the action of TET and TDG enzymes, and then re-written. In this scenario, it is interesting that blocking TDG appeared to promote the cell cycle rate, and could suggest that demethylation is a rate limiting step in cell cycle progression to ensure proper methylation of DNA in both daughter cells.
Because of the difference in expression levels of TDG between pluripotent and tissue derived NPCs (Figure 1A), we expected that silencing TDG would have a positive effect on the progression of developmental maturity of the NPCs. The LIN28/let-7 circuit was previously shown to be differentially regulated between NPCs born from pluripotent stem cells versus those derived from tissues, and resolution of this discrepancy was sufficient to advance the developmental maturity of NPCs in that context17. When the expression of TDG was brought down to a level similar to that seen in tissue derived NPCs, instead of advancing developmental maturation, the cells appeared to be unable to efficiently differentiate (Figure 2). This was presumably due to the increased rate of proliferation of the NPCs, a cell type where forced exit of the cell cycle is known to induce differentiation. Therefore, experimentally regulating TDG levels does not facilitate differentiation from pluripotent stem cells, as was the case with experimental downregulation LIN28. Perhaps the more interesting result from this work is that pluripotent derivatives probably need to silence TDG expression or activity at a more developmentally appropriate time point to proceed through proper development.
H9 hESCs and XFIPS2 were used in this study in accordance with the UCLA Embryonic Stem Cell Research Oversight committee (ESCRO, 2006-019-11A) and the Institutional Biosafety Committee (IBC). This work was specifically described in our ESCRO application (2006-019-11A), and approved on an annual basis by the committee. This work is considered not human subjects research by the UCLA IRB.
Cells were cultured in feeder free conditions on Matrigel (Corning) using mTeSR1 (Stem Cell Technologies) and passaged mechanically or with collagenase every 4–5 days. NPCs differentiation was performed as described previously (10). Briefly, for rosette induction, 80% confluent hPCS were cultured in DMEM/F12 with N2 and B27 supplements (Invitrogen), 20 ng/ml basic fibroblast growth factor (bFGF; R&D Systems), 1 µM retinoic acid (Sigma), 1 µM Sonic Hedgehog Agonist (Purmorphamine; Sigma), 10µM SB431542 (TGFß inhibitor; Cayman) and 0.1µM LDN193189 (BMP receptor type 1 inhibitor; Cayman). Small molecules were resuspended according to each manufacturer instructions. After about a week of culture neural-like rosettes were mechanically picked and expanded in NPC maintenance medium: DMEM/F12 supplemented with N2/B27, bFGF and 500 ng/ml epidermal growth factor (EGF; GIBCO). For further differentiation, the growth factors bFGF and EGF were withdrawn from the media (GFW) and cultured for 3 weeks. TDG and control knockdown in NPCs was performed using a unique 27-mer siRNA duplexes (Trilencer, Origene) at a final concentration of 20 nM using Lipofectamine RNAiMAX transfection reagent (Invitrogen).
Immunofluorescent staining was performed using standard protocol10,17. Briefly, cover slips were fixed with 4% PFA in PBS for 20 min, washed and then permeabilized and blocked in 10% donkey serum, 0.01% Triton in PBS for 1 hour. Primary antibodies in 5% donkey serum were incubated for 1–2 hours at room temperature following 3X wash in PBST and incubation with conjugated secondary antibody for 1 hour in room temperature. After 3X wash with PBST cover slips were incubated with 300nM DAPI final concertation in PBST for 3 min in room temperature (dark), followed with 3X wash with PBST. Antibodies used include the following: polyclonal rabbit anti-TDG 1:100(Atlas; HPA052263); polyclonal chicken anti-GFAP 1:1000(Abcam; ab4674); mouse monoclonal anti-MAP2 1:500 (Abcam, ab11267), polyclonal rat anti-KI67 1:100 (eBioscience; 14-5698). For methylation modifications, permeabilized cells were denatured with 2N HCl for 15 min and then neutralized with 100 mM Tris-HCl (pH 8.5) for 10min before blocking. The following Active-Motif Antibodies were used: rabbit anti 5hmC 1:100(39770); rabbit anti-5fC 1:2500 (61223); rabbit anti-5caC 1:1000 (61225). Image analysis and quantification was performed using ImageJ version 1.50i with the same threshold for each channel for all samples. Western Blot analysis was performed using standard procedures as described (Lowry et al., 2005) antibodies used were rabbit anti-TDG as described above and mouse anti-Actin 1:1000 (Santa Cruz Biotechnology, sc-47778).
RT-PCR Analysis. Total RNA was extracted using an RNeasy Mini Kit (QIAGEN). cDNA synthesis was performed using the Superscript III first-strand cDNA synthesis kit (Invitrogen). Real-time PCR was performed in triplicate using the SYBR green real-time PCR MIX (Roche) in the Roche lightcycler 480 machine. Run was performed for 50 cycles and analysis was performed in Microsoft Excel 2013 using the 2-ΔΔCT method.
Total RNA was extracted using an RNeasy Mini Kit (QIAGEN). Libraries were constructed according to manufacturer instructions (TruSeq Stranded Total RNA with Ribo-Zero; Illumina). Following second strand PCR amplification, ~200bp sized libraries were excised from agarose gel and pooled together in 10mM concentration each. Samples were sequenced using Illumina HiSeq2000 on single-end 50-bp reads and aligned to human reference genome (Hg19) using Tophat (version 2.0.6)18. Processing using Cufflinks and Cuffdiff was performed to obtain differential fragments per kilobase of transcript per million mapped reads (FPKM)18. Three biological replicates (i.e. 3 separate knockdown experiments in different PSC clones) were grouped together. Further analysis was performed using the cummeRbund suite (v2.0.0). Functional annotation was performed using DAVID (V6.7)14.
Following trypsin dissociation, knocked-down NPCs were fixed overnight in 70% ethanol at -20°C. Fixed cells were then stained for half an hour at room temperature in the dark, with Propidium Iodide (PI) for a final concertation of 50 µg/ml supplemented with RNAse (final 1 µg/ml). DNA content was analyzed on BD-Biosciences LSR-II flow cytometer and cell cycle phases were determined using the FlowJo cell cycle module (version 7.6.5).
Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen; 69506). One µg genomic DNA was treated by M.SssI (New England Biolabs; M0226s) in a 50 µl reaction for three rounds. For each round DNA was incubated with 4 Units of M.SssI CpG methyltransferase (NEB), supplemented with 160 mM final S-Adenosyl methionine for 3 hours at 37°C. At the end of each round DNA was cleaned using phenol/chloroform extraction. Bisulfite conversion was performed using the EpiTect Bisulfite Kit (QIAGEN; 59104) and then selected loci was PCR amplified with KAPA HiFi Hotstart Uracil+ DNA polymerase (KAPABiosystems). The resulting PCR product was cloned into the TOPO-Blunt (Invitrogen) vector, and sent to Laragen for Sanger sequencing (GeneWiz). Analysis and visualization of sequence reads was done using the online BISMA tool.
The FUCCI reporter lentiviral plasmids, pCSII-EF-mCherry-hCdt1(30/120) and pCSII-EF-mVenus-hGeminin(1/110) were a generous gift of Dr. Atsushi Miyawaki (RIKEN Brain Science Institute, Saitama, Japan). Lentiviral virions were generated in 293T cells using standard protocols as previously described13 followed by concentration with Amicon Ultra-15 centrifugal units (100K; Millipore). hPSC were single celled with TryplE (Thermo) and re-plated 24h prior to infection supplemented with 10 µM Rho-associated kinase (ROCK) inhibitor Y27632 (Stemgent). Cells were first infected with one reporter lentivirus particles for overnight infection. Cells were washed with fresh medium and grown for 2– 3 passages for recovery and expansion. Next, we FACS sorted using FacsARIA (Becton Dickinson) the cells for the corresponding reporter to ensure that all cells are infected. Briefly: cells from a whole 6 well plate were treated with ROCK inhibitor for one hour, single celled using trypLE and re-suspended in PBS. After sorting cells were a re-plated for recovery for 5–7 days and then
We infected the cells with the second reporter using that same procedure followed by a second FACS sorting for the reporter.
Student’s t-test was performed using GraphPad 6.01. Results were judged to be significant if the p-value was < 0.05. All other statistical analysis described in this study were performed using Microsoft Excel 2013
Dataset 1: TDG regulates cell cycle progression in human neural progenitors. Complete dataset of all underlying data divided into folders based on relevant figures. 10.5256/f1000research.13801.d20137919
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)