Keywords
Zaire ebolavirus, viral protein 24, terpenoid, pharmacophore, molecular docking simulation, ADMET.
This article is included in the Cheminformatics gateway.
Zaire ebolavirus, viral protein 24, terpenoid, pharmacophore, molecular docking simulation, ADMET.
The Ebola virus disease (EVD) is an often-lethal hemorrhagic fever for human and non-human primates. The World Health Organization (WHO) has declared EVD one of the global public health threats. EVD is caused by various Ebola virus species that are members of the Filoviridae family, some of which have spread to countries like the United Kingdom and the United States1. Five Ebola virus species cause Ebola disease, namely Bundibugyo, Reston, Zaire, Tai Forest, and Sudan2. The Zaire ebolavirus (EBOV) is the most virulent among them, with a mortality rate of up to 90%, and is responsible for all EVD outbreaks that have occurred worldwide since 1976, mainly in Gabon, the Democratic Republic of the Congo, and West Africa1. The symptoms of EVD include myalgia, fever, decreased plasma volume, anorexia, hypotension, diarrhea, and mucosal bleeding in the genitourinary and gastrointestinal tracts2.
EBOV’s genome includes seven genes that encode as many proteins that play essential roles in the lifecycle of the virus. The seven genes are known to encode the viral proteins (VP) 24, VP30, VP35, VP40, nucleoprotein (NP), glycoprotein (GP), and L (polymerase)1. Of importance amongst these, VP24 is responsible for the maturation of viral nucleocapsids and the regulation of viral transcription and replication. VP24 and VP40 have important roles in virus budding and assembling, the VP24 and NP have a contribution for viral budding and releasing3. VP24 also has a key role in the inhibition of the host cells’ immune system, especially regarding interferon signaling. VP24 is able to bind karyopherin alpha, which prevents the accumulation of tyrosine-phosphorylated STAT1 (pSTAT) in the nucleus. Afterward, the binding of IFN-stimulated response elements (ISREs) does not occur and inhibits the interferon signaling system and causes inhibition of host-mediated IFN responses as the body's defense against the Ebola virus4. Therefore, VP24 is a potential drug target in the treatment of EVD5.
Until now, no drugs have been developed to treat EVD, and potential candidates are still in the development stage. Computer-aided drug discovery and development (CADDD) is an efficient approach to identifying compounds characterized by high affinity and selectivity for macromolecular targets and beneficial pharmacokinetic and pharmacodynamic properties6. Use of CADDD enables the screening and prediction of the absorption, distribution, metabolism, excretion, and toxicity (ADME-Tox) of drug candidates for drug discovery and development7. Therefore, with CADDD we were able to quickly eliminate drug candidates with low biological activity and unsatisfactory ADME-Tox properties, avoiding the implementation of the time-consuming subsequent steps of the analysis7. The pharmacophore features test in CADDD enables identification of molecular species that are likely to display a high inhibitory affinity and interaction with macromolecules recognized as important targets for disease treatment8. The site of the pharmacophore is indicated with vectors and colored spheres that denote the atom’s characteristics: hydrophobic, hydrogen-bond acceptor, hydrogen-bond donor, aromatic, cationic, or anionic9. Pharmacophore features are an advanced tool in drug discovery to screen drug candidates, optimize lead compounds, and design new compounds. Additionally, they are commonly used as templates to screen molecules (hits) with similar chemical features8.
Terpenoids are a group of compounds that we investigated as inhibitors of EBOV VP24 via computational pharmacophore-based drug design. Terpenoid compounds are the largest group of natural products, and they are built starting from isoprene units and synthesized through the mevalonate and non-mevalonate pathway10. Members of this compound family are classified according to the number of isoprene units that make up their structures; namely, hemiterpenoids comprise one isoprene unit and five carbon atoms, monoterpenoids are composed of two isoprene units and ten carbon atoms. Sesquiterpenoids have three isoprene units and fifteen carbon atoms, diterpenoids have four isoprene units and twenty carbon atoms, sesterterpenoids have five isoprene units and twenty-five carbon atoms, triterpenoids have six isoprene units and thirty carbon atoms, tetraterpenoids have eight isoprene units and forty carbon atoms, and polyterpenoids have more than forty carbon atoms and more than eight isoprene units10. Terpenoids exhibit unique pharmacological and biological activities, including antibacterial11, anti-inflammatory12, anticancer13, and antiviral14 activities.
This study was performed employing a CADDD approach using the pharmacophore feature to identify pharmacological properties and inhibitory activity toward EBOV VP24. Various online and offline software packages, such as Molecular Operating Environment 2014.09 (MOE), Toxtree v2.6.13, DataWarrior v04.07.02, AdmetSAR, pkCSM, and SwissADME, were used to analyze the pharmacological properties of all ligands and to obtain the best drug candidates for anti-EVD therapy15,16. Figure 1 presents the research pipeline of this study.

The 3D crystal structure of EBOV VP24 was obtained from the RCSB Protein Data Bank —PDB ID: 4M0Q by Edwards et al.17. Data on 4M0Q were saved in .pdb format, and the file thus obtained was then opened in MOE 2014.09 for protein preparation and optimization. In this study, preparation and optimization of 4M0Q was performed using the LigX features in MOE 2014.09 with the Amber 10: EHT and solvation of the gas phase. Notably, AMBER10: EHT has been chosen as the forcefield for simulations involving large biomolecules, like proteins and nucleic acids18. Then, the geometry of the protein crystal was optimized, and the energy of the protein was minimized. Finally, the optimized structure of EBOV VP24 was stored in .moe format.
Terpenoids were obtained from the PubChem database. They are classified as organic chemicals in the chemicals and drugs category. In the organic chemicals category, terpenoids belong to the hydrocarbon group. Data from terpenoids from several subclasses, namely cannabinoids, carotenoids, diterpenes, dolichol, gefarnates, hemiterpenes, monoterpenes, polyisoprenyl phosphates, sesquiterpenes, sesterterpenes, and triterpenes, were collected. The structures of 55,979 terpenoids were saved in .sdf format.
The mentioned structures were evaluated performing the toxicity prediction test using DataWarrior v04.07.02 to identify ligands with favorable characteristics for molecular docking. Subsequently, the ligands underwent preparation and optimization with MOE 2014.09 software using modified MMFF94x as a force field and gas phase solvation. The standard ligands in this stage of the study were gossypetin and bisdemethoxycurcumin which were used to compare the characteristics of the best drug candidates for treating the EVD19. Thus, the structures of the ligands selected were saved in .mdb format.
In drug discovery, the determination of pharmacophore features contributes to the identification of ligands that have the potential to achieve a specific desired effect on a target molecule. The pharmacophore features of EBOV VP24 were obtained through molecular docking simulations with standard ligands. The standard ligands were made to dock using the site finder feature, which enabled us to identify the binding pocket of 4M0Q/EBOV VP24. Then, the molecular interaction of standard ligands with EBOV VP24 was used to determine the protein’s pharmacophore features through the pharmacophore query editor in the MOE 2014.09 software with gas phase solvation and Amber10: EHT as a force field. Finally, the pharmacophore features of 4M0Q/EBOV VP24 were saved in .ph4 format20.
A two-step molecular docking simulation was performed: rigid docking followed by flexible docking. The rigid docking process refers to the condition whereby the protein is regarded as rigid and the ligand as dynamic. Therefore, the ligand will find the most stable conformation with the protein target without the latter undergoing any changes in bond angles or bond lengths. On the other hand, in the flexible docking protocol, the conformational change that the binding site of the protein target undergoes upon docking is also taken into consideration. Ligand and protein are regarded as flexible in this case, and they will seek the conformation of the most stable ligand-protein complex to optimize the strength of the bonds between ligand and receptor21. In this study, the rigid and flexible molecular docking simulations were used alongside the pharmacophore features to dock the ligands to 4M0Q. Molecular docking simulations were performed using MOE 2014.09 software with gas phase solvation and AMBER 10: EHT as a force field.
Two types of scoring functions were used in our study, namely London dG and GBVI/WSA dG. The London dG scoring function provides analytical data on hydrophobic, ionic, and hydrogen-bonding interactions and affords an estimate of the free binding energy of ligand-receptor bonds based on the given conformations22. The GBVI/WSA dG scoring function enables researchers to determine the value of refinement parameters that will afford an estimate of the ligand binding energy in each pose. Moreover, the GBVI/WSA dG scoring function displays high sensitivity to the modification of the protein environment. Therefore, docking accuracy and the strength of protein-ligand bonds can be influenced by the GBVI/WSA dG scoring function23. Based on the results of molecular docking simulations, ligands were dropped from further analysis when they displayed values for Gibbs free binding energy (∆Gbinding) that were smaller than those of the standard ligands and when the value of the root-mean-square deviation (RMSD) of the ligands was above 2.0 Å.
The best ligands based on the results of the previous analysis underwent pharmacological property analysis. Various online and offline software packages were used to screen the ADME-Tox properties of the ligands, such as DataWarrior, Toxtree, SwissADME, admetSAR, and pkCSM. DataWarrior was used to analyze the ligand’s mutagenicity, drug-likeness, and tumorigenicity. Toxtree was used to evaluate the carcinogenicity and mutagenicity of the ligands24. SwissADME was utilized to predict ligand’s pharmacokinetics and physicochemical properties25. Finally, the absorption, distribution, metabolism, and excretion properties of the ligands were predicted with admetSAR and pkCSM26.
VP24 is an essential protein in EBOV’s lifecycle. Therefore, VP24 is a potential target when trying to inhibit EBOV replication and prevent EVD progression. The protein with PDB ID: 4M0Q was chosen as EBOV VP24 crystal structure, based on the fact that this structure has also been used in other studies19. The structure of PDB ID: 4M0Q was obtained through X-ray diffraction with a resolution of 1.921 Å. The protein’s active site comprises several amino acid residues, namely Y41, G44, I45, E46, F47, D48, R154, S151, S155, L158, S225, T226, F230, and T23127. In this study, all non-essential molecules in EBOV VP24 structure with PDB ID: 4M0Q, such as water, were eliminated, because their presence in the crystal structure can affect the interactions between ligand and protein28. The protonate 3D step was used for the addition of hydrogen atoms to the protein structure to mimic the hydrogen atoms in protein structure that may occur in nature15.
In this study, 55,979 terpenoids were selected in the PubChem database as potential inhibitors of EBOV VP24. The ligands were screened using DataWarrior v04.07.02 to identify disqualifying properties, such as mutagenicity, tumorigenicity, having negative reproductive effects, and being an irritant, and to recognize positive traits like drug-likeness. In fact, the DataWarrior software can be used to predict reproductive effects as well as the mutagenic, irritant, and tumorigenic properties of a particular compound based on its constituent fragments. Drug candidates displaying similar or identical structural traits as compounds known to be an irritant, upon contact, can cause the human body to display dryness or reddening29. In the DataWarrior software package, benzanthrone derivatives, silicon ether, and halogenated phenol ethers are regarded as irritant structural alerts whose use can lead to irritation in the human body30. The tumorigenicity of drug candidates is another important trait to identify because tumorigenic compounds can induce tumor cell proliferation in human tissues31.
Compounds predicted to pose a risk due to their mutagenic characteristics may cause transmissible genetic damage, including gene mutations, chromosomal aberrations, and changes in the number of chromosomes32. Nitroaromatic compounds, nitrobenzene derivatives, and phenylamine derivatives are regarded as mutagenic structural alerts by DataWarrior33. In the present study, gossypetin was predicted to be mutagenic because of its 5,8-dihydroxychromone fragment. Based on toxicity predictions made using DataWarrior, 3,353 ligands were identified as possessing the desirable characteristics to inhibit EBOV VP24 activity. The ligands’ geometries were optimized to obtain ligands having the features that can be using the MMFF94x force field with gas phase solvation15.
Pharmacophore features play a vital role in determining the optimal molecular interactions between ligand and target macromolecule that trigger or block a particular biological activity of the macromolecule. Pharmacophore features are identified through ligand-based or structure-based construction. In the ligand-based construction, the pharmacophore feature is determined by collecting a set of ligands which shows biological activity against a particular target macromolecule and extracting the chemical properties that confer upon the molecules their bioactivity. In the structure-based construction, the pharmacophore feature is determined via analysis of the possible interaction sites between ligands and macromolecular targets. Thus, pharmacophore features identified can be used in de novo design, virtual screening, or for the optimization of lead compounds. In pharmacophore-based virtual screening, a particular pharmacophore feature is used as a screening template to find molecules that have a similar feature as the template (hits)8. Therefore, based on similarities with the pharmacophore feature used as a template, the hits are expected to display the desired biological activity.
In this study, two pharmacophore features were generated based on the interaction between the standard ligands (bisdemethoxycurcumin and gossypetin) and the protein target (4M0Q EBOV VP24)19,34. As shown from the structures of the ligand-target molecule complexes depicted in Figure 2, the hydrogen acceptor (acc) feature, reported in blue color, was constructed based on the interaction between standard ligands and the side chain of arginine B154. The hydrogen donor feature, reported in purple, was generated in reference to the interaction between the standard ligands and the side chain of aspartic acid A48. The pharmacophore features of EBOV VP24 were analyzed and validated using standard ligands. Finally, 1,375 hit ligands that match the pharmacophore features mentioned above were identified so that they could be used in molecular docking simulations. The 4M0Q EBOV VP24 protein along with the pharmacophore features is presented in Figure 2.

Pharmacophore features of Zaire ebolavirus protein VP24 (PDB ID: 4M0Q) identified by interaction with ligands (A) bisdemethoxycurcumin and (B) gossypetin located in the viral protein’s binding pocket
In this study, molecular docking simulations were performed to assess the protein-ligand binding affinity and predict the conformations of ligand and macromolecular target in the protein-ligand complex35. All of the 1,375 hit ligands identified in the previous stage of the procedure underwent a molecular docking simulation through pharmacophore-based rigid docking. In particular, a retain value of 30 was used to identify hits in the first simulation, and a retain value of 100 was used for the same purpose in the second simulation with Amber10: EHT as a forcefield and gas phase as the solvation parameter for both simulations. Of the 1,375 ligands whose structures were subjected to the pharmacophore-based rigid docking simulation, 855 were considered satisfactory as the values assigned to the relevant simulation at a minimum matched the retained value of 30. Then, these hit ligands were screened based on their RMSD value (<2 Å), leading to the selection of 651 ligands. These 651 ligands were then used in a pharmacophore-based rigid molecular docking simulation using a retain value of 100, leading to the selection of 642 ligands. Then, these ligands were screened based on their RMSD value, leading to the selection of 581 ligands. These 581 ligands were subjected to pharmacophore-based flexible docking simulations utilizing a retain value of 100. The 577 ligands were selected, and their RMSD values were evaluated, leading to the selection of 344 ligands, which were analyzed based on their ∆Gbinding value, RMSD value, and interaction with the 4M0Q EBOV VP24 protein. In particular, ligands were selected if the value of their ∆Gbinding was lower than the corresponding parameters calculated for the standard ligands, indicating that the relevant protein-ligand conformation was more stable than that involving the standard ligands and 4M0Q15. Rigid docking show the best performance rates between 50-75%, while flexible docking methods can enhance pose prediction up to 80–95%36.
Additionally, ligands were selected if their RMSD value was below 2.0 Å since a value of this magnitude indicates that the protein and ligand conformations produced by the molecular docking simulation are highly accurate37. In this study, the ability of ligands to inhibit the activity of the target protein was predicted through the pKi value. A large, positive value for pKi indicates that the protein-ligand complex has a low dissociation rate38. Then, all ligands were screened based on their interaction with 4M0Q. Ultimately, six ligands were selected as the best candidates for the inhibition of 4M0Q activity, namely 3-O-acetyluncaric acid, carnosic acid, ilexgenin A, negundoside tasumatrol G, and taxumairol V, which are characterized by advantageous traits regarding ∆Gbinding, RMSD value, and strength of the protein-ligand interaction.
Among these six inhibitor candidates, there are three ligands which fulfill the topological polar surface area (TPSA) value (in excess of 140 Å)39. Increased permeation rate are correlates to decreased polar surface area40. Carnosic acid displayed the lowest RMSD value among the six selected terpenoids, although standard ligand gossypetin had an even lower RMSD value than carnosic acid. Negundoside had the lowest ∆Gbinding value and the highest pKi value, indicating its superior ability to inhibit the activity of EBOV VP24. Table 1 summarizes the molecular properties of the six ligands just discussed and of the two standard ligands.
| Ligand Name | Flexible Docking | MW | pKi | TPSA | |
|---|---|---|---|---|---|
| ∆Gbinding | RMSD | ||||
*Bisdemethoxycurcumin | −6.560 | 1.862 | 308.332 | 4.779 | 74.60 |
*Gossypetin | −7.266 | 0.435 | 318.236 | 5.293 | 147.68 |
3-O-acetyluncaric acid | −8.009 | 1.169 | 472.556 | 5.834 | 130.36 |
Carnosic acid | −8.706 | 0.689 | 443.566 | 6.342 | 110.70 |
Ilexgenin A | −8.009 | 0.855 | 500.673 | 5.834 | 120.72 |
Negundoside | −9.861 | 1.123 | 495.455 | 7.183 | 195.27 |
Tasumatrol G | −9.704 | 0.842 | 630.684 | 7.069 | 175.12 |
Taxumairol Vv | −9.252 | 1.021 | 415.460 | 6.739 | 186.12 |
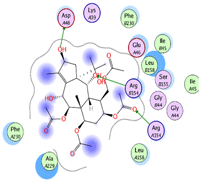
In this study, the molecular interactions between the target protein and ligand identified by molecular docking simulations were analyzed to quantify the ability of each ligand to interact with the target protein. All ligands were found to interact with the target protein’s active site. Ilexgenin A displayed the highest number of molecular interactions with the target protein; it displayed interactions with six active-site amino acid residues of EBOV VP24, namely glycine B44, isoleucine B45, glutamine B46, aspartic acid A48, arginine A154, and phenylalanine A230. Negundoside is the second-best ligand in terms of the number of interactions with the target protein. This ligand is involved in hydrogen-binding interactions with five amino acid residues in the protein’s active site: lysine A39, glutamic acid A46, aspartic acid A48, arginine A154, and phenylalanine A230.
Tasumatrol G is involved in three hydrogen-binding interactions with amino acid residues in the protein’s active site: aspartic acid A48, arginine A154, and alanine A229. Taxumairol V and carnosic acid are each involved in three hydrogen-binding interactions with EBOV VP24. Taxumairol V is involved in hydrogen-binding interactions with three amino acid residues located in the active site of EBOV VP24, namely aspartic acid A48, arginine A154, and arginine B154. Meanwhile, the carnosic acid showed have hydrogen-bonding interactions with aspartic acid A48, arginine A154, and arginine B154. The 3-O-acetyluncaric acid showed the lowest number of hydrogen-binding interactions with the target protein. Table 2 depicts the structures of selected simulated ligand–4M0Q complexes and the interactions that characterize them.
Pharmacological tests were carried out to predict the absorption, distribution, metabolism, excretion, and toxicity (ADME-Tox) properties of the six selected terpenoid ligands, and to identify the ligand among them with the potential to be used as a drug in EVD therapy (full data for the final 6 ligands is available as underlying data41. The prediction of ADME-Tox properties was performed using various software packages because each of them evaluates different traits in the pharmacological tests. In this study, the pharmacological tests were performed with software packages Toxtree, swissADME, pkCSM, and admetSAR.
Toxtree analysis was implemented to determine the mutagenic potential regarding Salmonella typhimurium TA100, potential carcinogenic based on the quantitative structure–activity relationship (QSAR), genotoxic carcinogenicity, and non-genotoxic carcinogenicity of the ligands. Table 3 summarizes the results of the pharmacological tests performed with Toxtree. According to this analysis, 3-O-acetyluncaric acid, taxumairol V, ilexgenin A, and negundoside were not potential carcinogenic, mutagenic, genotoxic carcinogenic, and non-genotoxic carcinogenic compounds. Carnosic acid exhibited the characteristics of a non-genotoxic carcinogen and a mutagen. Genotoxic carcinogens can interact directly with DNA, thus, producing DNA adducts and DNA damage. Non-genotoxic carcinogens, on the other hand, do not interact directly with DNA, but they can damage cellular structures and change cell proliferation rates42. The Toxtree software comprises several structural alerts for non-genotoxic carcinogens, including the presence of thiocarbonyl, halogenated benzene, and halogenated dibenzodioxin groups43. Carnosic acid and tasumatrol G exhibited the characteristics of non-genotoxic carcinogens because they include n-alkyl carboxylic acid and thiourea groups within their structure, which match the software’s thiocarbonyl structural alert. Thiourea is a non-genotoxic carcinogen that can cause cancer in the thyroid gland. In fact, thiourea acts as a peroxidase inhibitor in the thyroid gland, inhibiting thyroxine production. A reduced synthesis level of the thyroxine hormone causes, in turn, the pituitary gland to increase thyrotropin hormone secretion, resulting in an increased risk of thyroid cancer44.
| No | Ligand | Pharmacological Properties | |||
|---|---|---|---|---|---|
| Genotoxic | Non-genotoxic | Potential S.typhimurium TA 100 mutagenicity | Potential Carcinogenicity based on QSAR | ||
| 1 | 3-O-acetyluncaric acid | No | No | No | No |
| 2 | Carnosic acid | No | Yes | Yes | No |
| 3 | Ilexgenin A | No | No | No | No |
| 4 | Negundoside | No | No | No | No |
| 5 | Tasumatrol G | No | Yes | No | No |
| 6 | Taxumairol V | No | No | No | No |
| 7 | *Bisdemethoxy- curcumin | No | No | No | No |
| 8 | *Gossypetin | No | Yes | No | No |
In this study, the potential mutagenicity of ligands was determined using the Toxtree and admetSAR software packages through the Ames test and potential S. typhimurium TA 100 mutagenicity test. In this test, chemicals are analyzed in terms of their mutagenicity toward S. typhimurium, given that bacteria belonging to this species are highly sensitive to mutagenic compounds. In particular, in Ames tests, S. typhimurium bacteria modified so that they cannot synthesize histidine are exposed to potentially mutagenic compounds. These compounds may in turn cause mutations in the bacterial his-operon, a type of change that would be made evident by the ability of the modified bacteria to grow in culture media lacking histidine. In the prediction of the mutagenic properties of ligands, the Toxtree software packages comprise a structural alert for mutagenic compounds based on results from Ames tests; structures that set off this alert include aromatic amines and nitroarenes45.
Given that carnosic acid comprises an aromatic amine group, Toxtree-based analysis classified this ligand as potentially mutagenic. In fact, aromatic amines will undergo biotransformation in the human body that results in the formation of covalent bonds with guanine, which may, in turn, cause DNA mutations. Therefore, carnosic acid was removed from the list of potential drug candidates for EVD therapy. Meanwhile, based on the Ames test which was performed using admetSAR software, all the best ligands showed no Ames toxic, which means that it did not have the potential to be mutagenic compound46. Importantly, the results of mutagenicity analysis performed using Toxtree and admetSAR were different from each other because the two software packages use different databases to formulate predictions on the mutagenic potential of ligands.
The six EBOV VP24 inhibitor candidates and the two standard ligands underwent analysis for potential mutagenicity, acute oral toxicity, human intestinal absorption (HIA), and cytochrome P450 (CYP)-inhibiting activity using the admetSAR software package. Table 4 reports data on the relevant pharmacological properties. HIA is an important oral bioavailability indicator47, and it is given a positive (+) value when a compound is promptly absorbed in the human intestine, or a negative (-) one, when the compound has poor absorption characteristics in the human intestine. The only negundoside displayed a negative HIA value following analysis, meaning that this ligand has poor or weak absorptivity in the human intestine. Regarding acute oral toxicity, compounds subjected to analysis may fall into four categories: category I designates highly toxic compounds, category II designates moderately toxic compounds, category III designates compounds of low toxicity, and category IV designates non-toxic compounds48. Acute oral toxicity analysis performed with the admetSAR software package led to ilexgenine A, negundoside, and tasumatrol G being classified as highly toxic. 3-O-acetyluncaric acid, taxumairol V, and carnosic acid were instead classified as low-toxicity compounds. Ilexgenine A and negundoside were, thus, removed from the list of potential candidates for the inhibition of EBOV VP24 activity. Trasumatrol G was retained for analysis given its good molecular docking characteristics.
| No | Ligand | Pharmacological Properties | |||
|---|---|---|---|---|---|
| Ames-test Mutagenicity | HIA | Acute Oral Toxicity | CYP Inhibitor | ||
| 1 | 3-O-acetyluncaric acid | No | + | III | None |
| 2 | Carnosic acid | No | + | III | CYP450 1A2, 2C9, 2C19 |
| 3 | Ilexgenin A | No | + | I | None |
| 4 | Negundoside | No | - | I | None |
| 5 | Tasumatrol G | No | + | I | None |
| 6 | Taxumairol V | No | + | III | None |
| 7 | *Bisdemethoxycurcumin | No | + | III | CYP450 1A2, 2C9, 2C19, 3A4 |
| 8 | *Gossypetin | No | + | II | CYP450 1A2, 3A4 |
It is essential to identify CYP-inhibiting properties to develop a new drug. In fact, CYPs belong to an enzyme family that plays a key role in the metabolism of drugs, as its members catalyze the biotransformation of drugs in advance of their decomposition and excretion49. The AdmetSAR software package can be used to analyze the ability of ligands to act as inhibitors of several CYP isoforms that play a role in drug metabolism, namely 1A2, 2C9, 2C19, 2D6, and 3A4. In this study, carnosic acid, bisdemethoxycurcumin, and gossypetin displayed the potential to inhibit CYP 1A2 activity. Carnosic acid and bisdemethoxycurcumin displayed the potential of inhibiting the activity of CYP 2C9 and CYP 2C19. Bisdemethoxycurcumin and gossypetin displayed the potential to inhibit CYP 3A4 activity. Drugs that have the potential to inhibit the activity of various CYP isoforms may cause undesirable side effects, as they may cause drug metabolic disorders and the accumulation of potentially toxic drug metabolites50. Therefore, it was concluded that carnosic acid, gossypetin, and bisdemethoxycurcumin were not suitable candidates for the development of drugs for EVD therapy.
Ligands’ potential for hepatoxicity and hERG I-inhibiting activity were analyzed using the pkCSM software package. Table 5 summarizes results from this analysis. The human Ether-à-go-go Related Gene (hERG) is a selective potassium ion channel that regulates potassium ion transfer and is expressed in various cells and tissues, including cardiac myocytes, neurons, smooth muscles, and neuroendocrine cells51. Inhibition of hERG I can extend cardiac repolarization, which may result in ventricular arrhythmias, torsades de pointes, and sudden cardiac death52. Terfenadine, cisapride, astemizole, and grepafloxacin are drugs known to have inhibitory effects on hERG. I, and they have been withdrawn from the market because they can cause arrhythmias53. None of the ligands tested were predicted to display any hERG I-inhibiting activity. Regarding the potential for hepatoxicity, carnosic acid was predicted to cause damage to the liver, an organ that plays an important role in the metabolism of proteins, fats, carbohydrates, and toxins54. Therefore, carnosic acid was removed from the list of drug candidates for the inhibition of EBOV VP24 protein activity.
| No | Ligand | Pharmacological Properties | |||
|---|---|---|---|---|---|
| hERG I Inhibitor | Hepatoxicity | Bioavailability | Synthetic Accessibility | ||
| 1 | 3-O-acetyluncaric acid | No | No | 0.56 | 6.64 |
| 2 | Carnosic acid | No | Yes | 0.55 | 3.03 |
| 3 | Ilexgenin A | No | No | 0.56 | 6.52 |
| 4 | Negundoside | No | No | 0.11 | 5.79 |
| 5 | Tasumatrol G | No | No | 0.17 | 6.61 |
| 6 | Taxumairol V | No | No | 0.56 | 4.91 |
| 7 | *Bisdemethoxycurcumin | No | No | 0.55 | 2.59 |
| 8 | *Gossypetin | No | No | 0.55 | 3.29 |
The predicted bioavailability and synthetic accessibility of ligands were determined through analysis performed with the swissADME software package (Table 5). Bioavailability is the fraction of a drug that can be absorbed by the body and the concentration of that drug which reach systemic circulation according to the total dose of the drug55. The bioavailability prediction score of a compound ranges from 0 to 1. If the bioavailability value of a compound is less than 0.5 (<0.5), the compound is predicted to have low bioavailability. If, on the other hand, that value is above 0.5 (> 0.5), the compound is predicted to have high bioavailability56. Based on the results of the bioavailability test, negundoside is predicted to display the lowest bioavailability (bioavailability score: 0.11). Therefore, negundoside was removed from the list of candidate drugs that inhibit VP24 Zaire ebolavirus activity.
Synthetic accessibility (SA) is an important parameter in drug design because, in some cases, compounds designed computationally prove very difficult to synthesize57. The SA score ranges from a value of 1, indicating that the compound is very easy to synthesize, to a value of 10, indicating that the compound is very difficult to synthesize25. Based on the low SA score, carnosic acid is predicted to be the easiest compound to synthesize. 3-O-acetyluncaric acid and ilexgenin A have high SA scores, meaning that these ligands are predicted not to be easily synthesized.
Results from the molecular docking simulations and pharmacological tests conducted in this study indicate that taxumairol V is the best drug candidate to inhibit EBOV VP24 protein activity, based on its ∆Gbinding, RMSD value, molecular weight, predicted interaction with the target protein and predicted pharmacological properties. Taxumairol V’s molecular weight is under 500 Dalton, and this ligand has a high propensity to inhibit the activity of the target protein, even though it does not have the highest pKi value among all the ligands tested. Furthermore, taxumairol V is predicted to pose no risk as a potential mutagenic, tumorigenic, carcinogenic, or irritant drug. This compound is predicted to display no inhibitory activity toward CYP, or hERG I. Taxumairol V is also predicted to display good HIA, and it is characterized by an intermediate bioavailability score. Finally, taxumairol V has an average SA score, indicating that it should be moderately easy to synthesize.
In silico approaches aid drug discovery and the development of chemical species that can achieve a desired biological effect. A total of 55,979 terpenoids were screened based on their potential toxicity, resulting in the identification of 3,353 compounds predicted to display beneficial properties. Six best drug candidates that have good molecular properties and are predicted to display strong molecular interaction with the EBOV VP24 protein were subsequently identified after molecular docking simulations were performed. These ligands were then subjected to pharmacological analyses performed using various online and offline software packages. Based on its molecular properties, predicted molecular interaction with the target protein, and predicted pharmacological characteristics, taxumairol V was identified as the best overall drug candidate to inhibit EBOV VP24 protein activity. Therefore, taxumairol V has the potential to be the basis for a drug to be developed to treat Zaire ebolavirus-caused Ebola disease.
Ebola virus VP24 structure from Protein Data Bank, Accession number PBD:4M0Q: http://identifiers.org/pdb:4M0Q
Figshare: All Test Result of Molecular Docking Simulation. https://doi.org/10.6084/m9.figshare.8275079.v141
This project contains the following underlying data:
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.















Comments on this article Comments (0)