Keywords
BACE1, ginsenoside, molecular docking, inhibitor, Alzheimer´s disease
BACE1, ginsenoside, molecular docking, inhibitor, Alzheimer´s disease
Alzheimer's disease is a neurodegenerative condition causing changes in behavioral patterns and progressive cognitive deterioration, leading to dementia and diminished orientation and recognition abilities1,2. It has been estimated that more than 45 million individuals have been diagnosed with dementia worldwide and projections for prevalence in 2050 indicate that around 130 million people will develop these types of manifestations3,4.
At present, Alzheimer's disease leads to irreversible repercussions for the diagnosed population, which constitutes a problem at the public health level. Current treatments are based on the functioning of neurotransmitters such as acetylcholine, specifically as acetylcholinesterase inhibitors, preventing the degradation of this endogenous substance and favoring the stimulation of associated neurotransmitter response mechanisms5,6. However, these treatments are lacking and are inefficient at significantly prolonging quality of life for patients.
Therefore, we have studied the properties of products of natural origin such as ginseng (Panax ginseng), specifically major secondary metabolites such as ginsenosides7, which are extracted from different plant components. These molecules generally contain steroid rings and have varied biological responses, such as anti-inflammatory, anti-carcinogenic and cardiovascular effects and, recently, activities at the level of nervous regulation, with potential effects on epilepsy, Parkinson’s and Alzheimer’s8–10.
Therefore, in this study, promising molecules from structural variants of ginsenosides with potential capacity of bound to proteins involved in the development of Alzheimer's disease such as β-secretase (BACE-1) were identified using interaction studies, identification of affinities, determination of binding-free energies and in silico methodologies based on molecular docking.
A search for ginsenoside Rg1 (CHEMBL501637) analogues was performed using the CHEMBL database11. The name of the structure ginsenoside Rg1 (CHEMBL501637) was used to search the database. Subsequently, a filter was applied to the analogues to identify compounds with 80–90% similarity. The database provided information about the physical representation, activity charts, calculated properties and solubility of these compounds. Previous theoretical studies with ginsenosides have reported finding few structures upon screening. A preliminary set database of 50 analogue structures with the highest similarities was developed. In order to evaluate this first set of molecules, over half of the structures (27) were randomly selected using a random number generator (accession numbers for these molecules are provided as Underlying data and their structures as Extended data). Two drugs were selected as references, which were searched for using the ZINC Database12 and PubChem, then selected and saved in MOL2 format. The selected structures were Donepezil (ZINC accession number 597013), due to its use in pharmacological therapy for mild and moderate Alzheimer’s disease. Likewise, verubecestat (PubChem accession number 51352361) was developed as a candidate molecule for the selective inhibition of BACE1.
BIOVIA Discovery Studio software version 4.513 was used to correct the structures through the conversion of the format of the structure, addition of hydrogen atoms, neutralization of the charged groups, generation of ionization states, clean geometry and, finally, the conversion of the output file into its final MOL 2 format. Subsequently, the representative crystallographic structure of BACE1 (Accession number 1FKN, resolution of 1.9 Å) was obtained from Protein Data Bank (PDB)14. For the preparation of the receptor, the protein obtained in PDB format was prepared through the addition of hydrogen atoms, the elimination of solvent molecules (water) and the allocation of partial charges using the software packages UCSF Chimera version 1.1315 and BIOVIA Discovery Studio version 4.516.
The molecular docking was performed with AutoDock Vina 4.2.117, using a PyRx working interface18. Prior to molecular docking, a virtual screening of 27 ginsenoside analogues was carried out to determine the molecules with the best structural affinity and candidates for a potential inhibitory effect of BACE1, whereby analogues were minimized by universal force field and conjugate gradients using a default algorithm based on Open Babel tools. BACE1 and ginsenosides analogue molecules were represented in a grid space of x = 25 Å, y = 25 Å, z = 25 Å A using a universal force field. Subsequently, each docking simulation used a single ginsenoside analogue molecule and the software selected the top eight conformations with the largest change in free energy of binding, which were classified according to the energy level of each conformation and according to root-mean-square-deviation value obtained. The result of each docked molecule was determined according to the docking energy of the receptor-ligand complex. The top conformations of the structures with BACE1 were obtained in pdbqt format, using PyMOL software version 2.3.219 for its visualization and conversion into PDB format. BIOVIA Discovery Studio version 4.5 was used to determine interaction forces and amino acids.
Docking interactions between the ginsenoside analogues with the highest binding affinity with BACE1 are shown in Figure 1A–E. The six analogues show binding energies of ≥7,5 Kcal/mol, affinity values that are comparative to those of the reference molecules. Equally, reference drugs, inhibitors of BACE1 and of acetylcholinesterase (AChE), were used in the study are shown in Figure 2A and 2B. Different types of interactions were observed between analogues and the residues of the pocket formed in the BACE1 enzyme, such as van der Waals, H-bonding (conventional and carbon), π-sigma, alkyl and π-alkyl. Likewise, common amino acids involved in binding with ginsenosides analogues such as L30, D32, S35, Y71, T72, Q73, I110, D228, T231 and T232 were identified. The binding affinity of selected ginsenosides, the BACE1 inhibitor and the acetylcholinesterase inhibitor with BACE1 are shown in Table 1, where it can be seen that the compound CHEMBL451292, CHEMBL510371, CHEMBL503302, CHEMBL201643, CHEMBL448647 and CHEMBL508182, showed the highest energy affinity, with values of -9.6, 8.1, 7.6, 7.5, 7.5 and 7.5 Kcal/mol, respectively. On the other hand, the reference drugs showed energies of binding 9.8 Kcal/mol for verubecestat and 8.3 Kcal/mol for donepezil. The hydrophobic residues Q12, I110, G11, T231, T232, I118, L30, G34, S35, D32, and N233 were found to strengthen the interactions between BACE1 and ginsenosides analogues through van der Waals interactions. It was also found that hydrogen bonding interactions between the active site of the BACE1 enzyme and ginsenoside analogues involved residues D32, Q73, N233, G264, R307, R235, T72, F322, K321, T231 and T329.
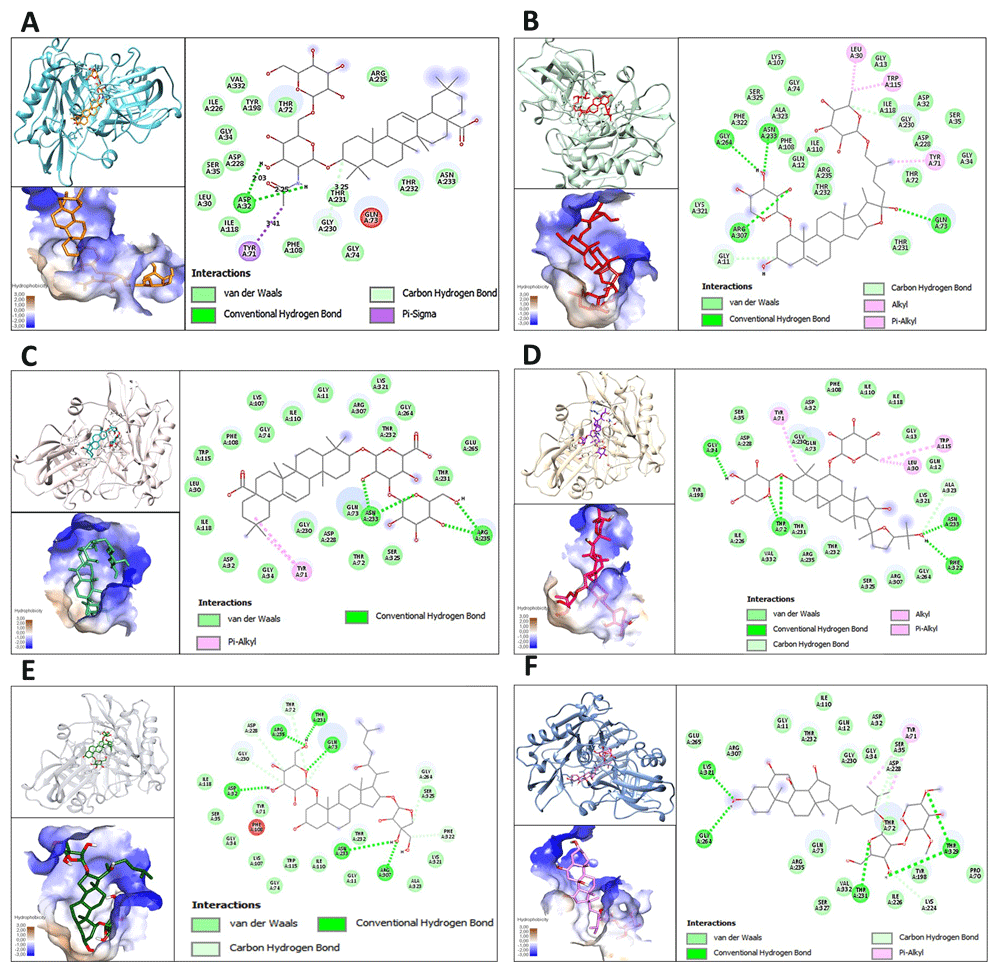
Molecular interactions of ginsenoside analogues and β-secretase 1 (BACE1 A. CHEMBL451292 analogue binding to BACE1. B. CHEMBL510371 analogue binding to BACE1. C. CHEMBL503302 analogue binding to BACE1. D. CHEMBL201643 analogue binding to BACE1. E. CHEMBL448647 analogue binding to BACE1. F. CHEMBL508182 analogue binding to BACE1.
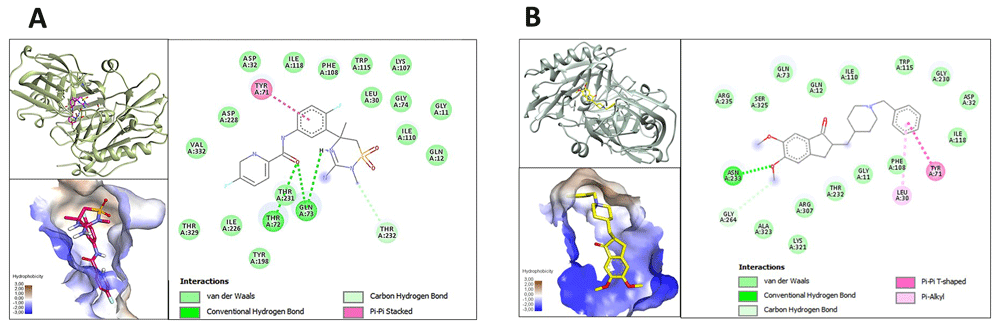
A. Verubecestat (inhibitor) binding to BACE1. B. Donezepil binding to BACE1.
It has been reported that ginsenosides demonstrate a variety of biological activities and due to this, have been progressively included as alternative in cardiovascular, antioxidant and anticancer therapies. Their recently discovered activities at the neuronal level establish a reason for the study of these metabolites in regards to neurodegenerative alterations such as Alzheimer's disease4,20, which are not fully understood at the molecular level. For example, the study of ginsenosides as anti-Alzheimer’s agents, specifically as potential BACE1 inhibitors21. Therefore, some authors have proposed experimental studies that indicate the potential role of these compounds as promising elements in the inhibition of BACE1 and as future molecules for the treatment of this pathology. Therefore, as an alternative to a molecular study and before the empirical approaches, in silico studies have been implemented to reveal the capacity binding and affinity of these metabolites and BACE1, studying the interaction and affinity energy of 27 randomly selected ginsenoside analogues.
The molecular docking experiments in this study revealed that the higher energy ginsenosides not studied previously demonstrate a considerable affinity with BACE1, specifically the analogues CHEMBL451292, CHEMBL510371 and CHEMBL503302, with binding energies of -9.6, -8.1 and -7,6 Kcal/mol, respectively. Amino acid residues such as D32, Q73, G230, N233, and R235 were identified as being associated with the formation of hydrogen bonds. Likewise, van der Waals hydrophobic interactions were established by residues L30, G34, S35, T72, F108, I110, I118, T231, T232, R307, K321 and S325, as reported in the study by Choi et al. 2016, which established a strong association between these interactions and the sugars of the molecular structures, which bear a great resemblance to the results obtained for the analogues with higher affinities22. On the other hand, similar interactions with the reference molecules (BACE1 and AChE inhibitors) with common residues identified in the present study and reported previously, such as D32, F108, I118, T232, N233 and K321, were demonstrated, with energy values of -9.8 and -8.3 Kcal/mol for the BACE1 and AChE inhibitors, respectively. It was indicated that the analogues CHEMBL451292 and CHEMBL510371, when interacting with BACE1, demonstrated a similar affinity to the reference drugs, with a higher affinity than donepezil and a similar affinity to Verubecestat, agents that are relevant to the current modern treatments against Alzheimer's disease. Donepezil is used in the usual treatment of the disease in its early and middle phases23 and, despite it having been established that its mechanism of action is based on the inhibition of AchE, our study established that said molecule exhibited binding to active site residues of BACE1, as shown in Figure 2B. On the other hand, Verubecestat is an experimental drug for the treatment of early stage disease, which was developed as an inhibitor of BACE1. During its development it showed interfering activity in the formation of the amyloid cascade and in avoiding the aggregation effect of the β-amyloid plaques. However, during the clinical phase, the study was discontinued due to safety concerns about observed alterations in the liver24,25.
Therefore, the use of computational tools for screening of new therapeutic agents with potential pharmacological activity could contribute to the elucidation at the molecular level of the mechanism of action of ginsenosides, as a group of promising metabolites for the prevention and treatment of Alzheimer's disease.
Figshare: Ginsenoside analogs as possible BACE1 inhibitors. https://doi.org/10.6084/m9.figshare.8408549.v126
This project contains the following underlying data:
Figshare: Ginsenoside analogs as possible BACE1 inhibitors. https://doi.org/10.6084/m9.figshare.8408549.v126
This project contains the following extended data:
- 597013_donepezil_out.pdb (donepezil structure in PDB format)
- 51352361_uff_E=743.53_out.pdbqt (verubecestat structure in PDBQT format)
- Structures of the 27 ginsenoside analogues in PDB format
Data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)