Keywords
influenza virus, pandemic, swine flu, mutations, electronic biology
influenza virus, pandemic, swine flu, mutations, electronic biology
Each flu season represents a serious public health threat and the challenge for health workers, vaccine producers and researchers due to unpredictable behavior of influenza A viruses. Vaccination is the most effective way to protect against seasonal influenza viruses. As a consequence of the high variability of influenza viruses, it is difficult to select vaccine candidates or predict the vaccine effectiveness (VE) for the upcoming season. Accordingly, the VE against the dominant A strain H3N2 in Australia in 2017 was very low (about 10%), and this country experienced record-breaking numbers of influenza-related hospitalizations and deaths (http://www.health.gov.au/internet/main/publishing.nsf/Content/cda-ozflu-2017.htm). The World Health Organization (WHO) selected the same vaccine strains for the United States for the flu season 2017–2018 and based on data coming from Australia 6 months before the beginning of the flu season, a similarly low VE against the H3N2 viruses was anticipated. This prediction of the potentially low VE in the USA, which was made by leading vaccine experts1, and aggressively promoted by press media, resulted in a very low vaccination rate in the USA. As a consequence, according to the CDC, 2017-2018 was the worst flu season on record, with an estimated 79,000 deaths, including total of 185 pediatric deaths. This number exceeds the previously highest number of flu-associated deaths in children reported during a regular flu season (171 during the 2012-2013 season). Approximately 80% of these deaths occurred in unvaccinated children.
Previous comparison of H3N2 viruses from 2017 from Australia with viruses collected during the pre-flu season 2017–2018 in the USA was performed using the electronic biology platform “wEB”, developed using methods described previously2. Our analysis demonstrated significant differences between these two groups of viruses3 allowing us to correctly predict higher VE against H3N2 virus for the flu season 2017–2018 in US than in Australia3. This prediction was in contrast with the prediction of VE of 10% based on Australian data1, and it was confirmed at the end of the flu season. CDC officials reported an overall VE of 36%, with VE of 25% against the H3N2 strain. The Armed Forces Health Surveillance Branch Air Force (AFHSB-AF) found that the vaccine provided substantially better protection to military dependents treated at the U.S. Air Force School of Aerospace Medicine, with an overall adjusted VE of 51% and a VE of 37% against H3N2. The Naval Health Research Center (NHRC) determined that the vaccine had even greater effectiveness among the civilians who received care for febrile respiratory illnesses, with an overall adjusted VE of 55% and VE of 52% against H3N2.
In contrast to the flu season in 2017–2018, which was dominated by H3N2 strains, the 2018–2019 flu season in the USA is heavily dominated by circulating influenza A(H1N1)pdm09 viruses between October 2018 and January 2019 (to access this data, visit https://gis.cdc.gov/grasp/fluview/fluportaldashboard.html and select Season 2018–19). This change in the dominant flu subtype was followed by an early beginning of the flu season and increased hospitalization and death rates for children and healthy young people in the beginning of the flu season for 2018–2019.
Herein, we analyzed sequences of HA of viruses A(H1N1)pdm09 collected in the USA during months of October and November of 2018 using our previously described electronic biology platform (wEB). The most recent analysis revealed that some of the (re)emerging viruses contain mutations previously identified as pandemic markers4. Moreover, the percentage of the viruses belonging to this population is significantly higher in October–November 2018 than in all past nine years. We also demonstrate that most of A(H1N1)pdm09 viruses have changed informational properties previously proposed to correlate with their biological and immunological properties. Possible consequences of these findings are discussed.
We analyzed the hemagglutinin subunit 1 (HA1) of (i) 33197 A(H1N1)pdm09 viruses collected between May 2009 and December 2017, (ii) 224 human pH1N1 viruses collected in US from October to November 2018 (GISAID database) and 28 human pH1N1 viruses collected in Canada during the same period and (iii) vaccine virus A/Michigan/45/2015 for 2018/2019 flu season. All sequences were stored in the publicly open database GISAID.
The ISM is a virtual spectroscopy method for the study of the long-range protein-protein interaction. According to this method, described in detail elsewhere2, sequences (protein or DNA) are transformed into signals by assignment of numerical values of each element (amino acid or nucleotide). These values correspond to electron-ion interaction potentials, determining the electronic properties of amino acid/nucleotides for their intermolecular interactions. The signal obtained is than decomposed in periodical function by Fourier transformation. The result is a series of frequencies and their amplitudes. The obtained frequencies correspond to the distribution of structural motifs with defined physico-chemical characteristics responsible for biological function of the sequence. When compared, proteins that share the same biological or biochemical function(s) possess the code/frequency pairs specific for their common biological properties. The method is insensitive to the location of the motifs and does not require previous alignment of the sequence.
The ISM served as a base for development of the phylogenetic algorithm for the Informational Spectrum-based Phylogenetic Analysis (ISTREE)5.
The phylogenetic tree of the HA1 influenza proteins is generated with the ISM-based phylogenetic algorithm ISTREE, previously described in detail elsewhere5. In the presented analysis, we calculated the distance matrix with the distance measure between sequences X1 and X2 defined as:
where A1(F1) and A2(F1) are amplitudes on frequency F1=0.295; A1(F2) and A2(F2) are amplitudes on frequency F2=0.055 in informational spectra on sequences X1 and X2 respectively.
Previously, we showed that the IS frequency F(0.055) of HA1 is responsible for interaction between H1N1/N2 and swine protein(s)/receptor(s) while the IS frequency F(0.295) for the same HA subunit is responsible for the interaction with human protein(s)/receptor(s)4. This suggests that the acquisition of mutations in HA1, leading to increased ratio of these amplitudes at these two frequencies A(0.29536)/A(0.055), are essential for adaptation of swine A/H1N1 viruses to humans, and this is currently used for real-time monitoring of A/H1N1 viruses. It has been also suggested that positions 94, 196 and 274 in HA1 of A(H1N1)pdm09 are hotspots for advantageous mutations for human adaptation of A(H1N1)pdm094. In Figure 1 and Table 1 we present the fraction of A(H1N1)pdm09 viruses with mutations in these hotspots that were collected between May 2009 and December 2017, and viruses isolated in between October and November 2018 (this period corresponds to the beginning of the flu season in the USA). Of note is that all of these mutations in 2018 appeared in A(H1N1)pdm09 viruses collected in October and November of 2018 in the USA.
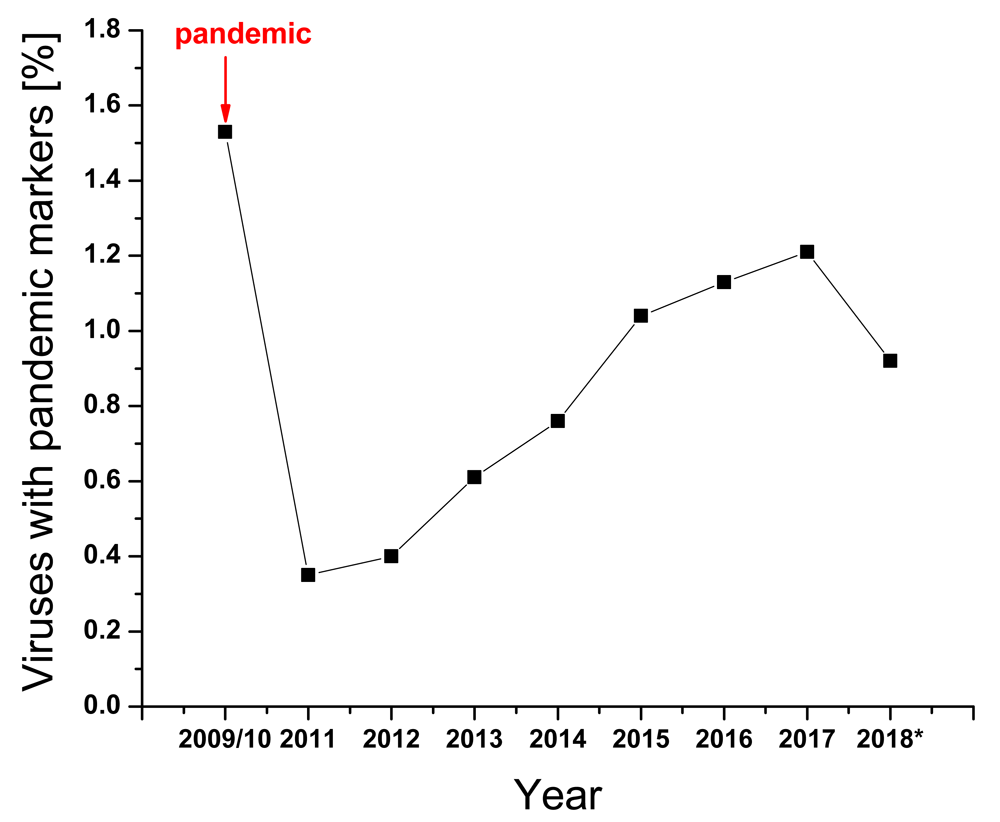
| Year | Total number of viruses | Viruses with markers | [%] |
|---|---|---|---|
| 2009/2010 | 11524 | 177 | 1.53 |
| 2011 | 1958 | 7 | 0.35 |
| 2012 | 990 | 4 | 0.40 |
| 2013 | 2305 | 14 | 0.61 |
| 2014 | 1964 | 15 | 0.76 |
| 2015 | 3267 | 33 | 1.04 |
| 2016 | 7159 | 77 | 1.08 |
| 2017* | 3464 | 42 | 1.21 |
| 2018 (Oct/Nov) | 566 | 9 | 1.59 |
| 2018 (Oct/Nov) USA | 244 | 9 | 3.69 |
In Figure 2a we present the consensus informational spectrum (CIS) of A(H1N1)pdm09 viruses isolated in the period of September–November 2018 in the USA. The dominant peak in this CIS corresponds to the frequency F(0.281). Viruses collected in the same period in Canada are characterized with the IS frequency F(0.295) (Figure 2b), which was previously identified as the hallmark of pandemic A(H1N1)pdm094.
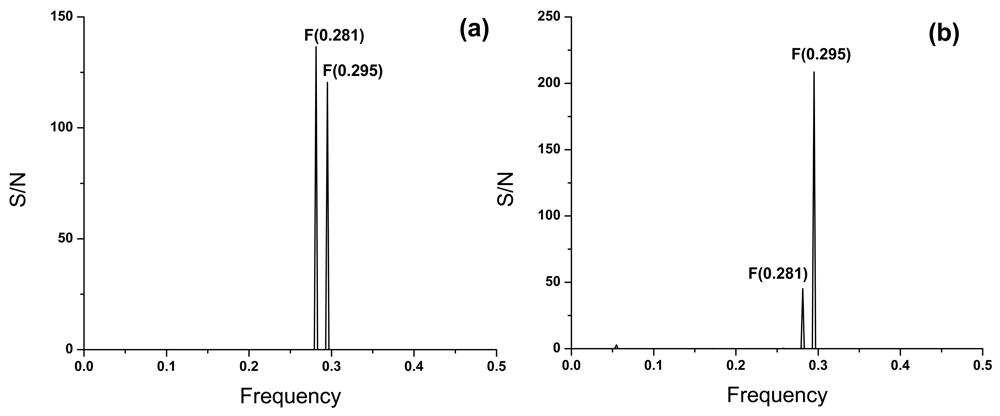
The consensus informational spectrum of A(H1N1)pdm09 viruses with pandemic markers which are collected in October and November 2018 in (a) the USA and (b) Canada.
Previously, we showed that the amplitude on characteristic frequencies in IS of HA1 from influenza A viruses is indicative of the vaccine efficacy against these viruses3,6. Accordingly, in Figure 3 we present the ISM-based phylogenetic tree of 224 A(H1N1)pdm09 viruses isolated in October and November of 2018 in the USA and the vaccine virus A/Michigan/45/2015. As presented, 101 (45 %) viruses are co-cauterized with the vaccine virus suggesting that the current vaccine can at least efficiently protect against this fraction of analyzed viruses.
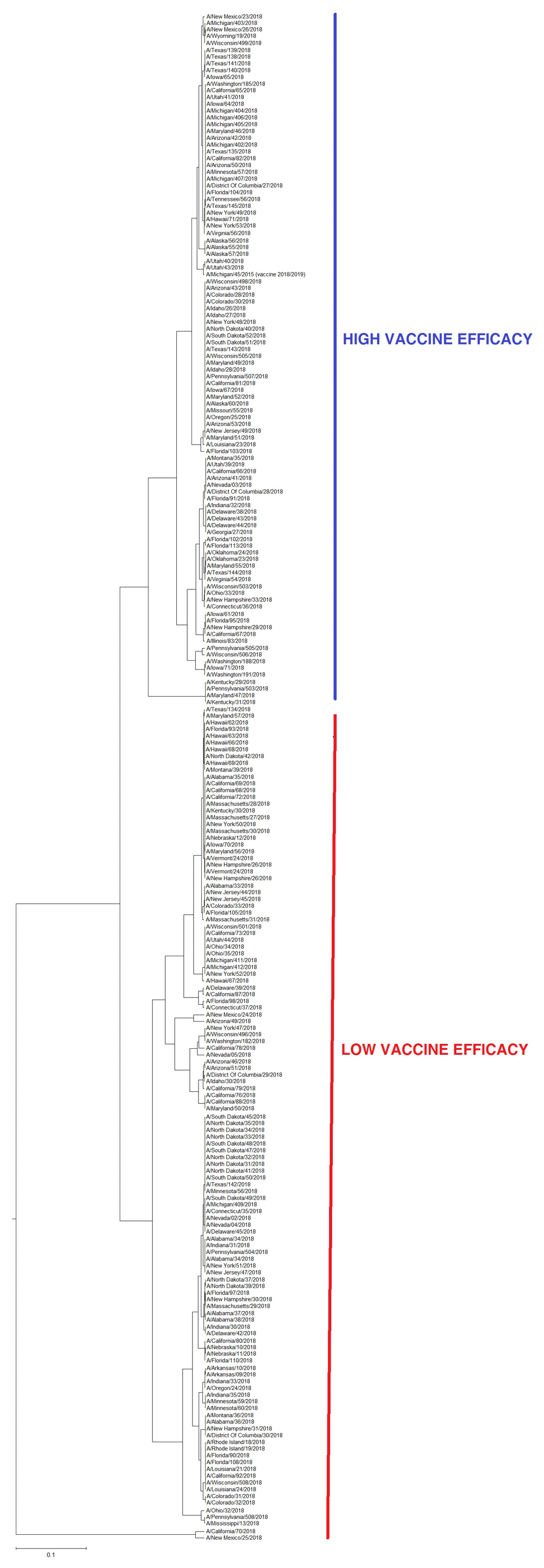
We previously showed that the ratio of amplitudes on the characteristic frequencies in IS of HA1 of influenza viruses determines adaptation of animal viruses to human7,8. In silico analysis of pandemic A(H1N1)pdm09 viruses revealed positions 94, 196 and 274 in HA1 as hotspots for mutations, which could increase infectivity of these viruses4. At the very beginning of the last pandemic, when this analysis was performed (May 2009), only three viruses from three countries (Spain, Italy and USA) in the GISAID database contained the mutations in these hot-spots. At the end of the pandemic (late 2010) sequences of 350 viruses with these mutations were deposited in GISAID. This suggests better adaptation of some viruses to humans during pandemic. The fraction of the viruses with these pandemic markers sharply decreased at the end of the pandemic in 2011. However, this viral population continued to slowly grow until 2017 while reaching the same end-pandemic level of 2010 in October and November of 2018 (Figure 1 and Table 1). It is also important to note that all viruses with pandemic markers isolated in October and November of 2018 are from the USA only, and that the fraction of these viruses is significantly higher than at the end of the 2009 pandemic (Table 1). This is a warning sign that the pandemic potential of A(H1N1)pdm09 viruses in the USA has increased. Of note also is that 7 of 9 of these viruses are isolated in patients 5 to 13 years old, suggesting that children could be more susceptible to infection with these viruses than adults.
Each subtype of influenza A viruses is characterized with specific frequency9. Results presented in Figure 2 show that HA1 from US viruses collected at the beginning of the 2018/2019 flu season are characterized with the IS frequency F(0.281). On the contrary, all other A(H1N1)pdm09 viruses isolated in the last 10 years are characterized with IS frequency F(0.295). This strongly suggests possible changes in the interaction profile of these viruses with host proteins. For now, it is not possible to predict how this change will influence pathological and immunological properties of A(H1N1)pdm09 virus.
Results presented in Figure 3 demonstrated that 101 of 224 (45%) A(H1N1)pdm09 viruses isolated in October and November 2018 in the USA are clustered in the ISM-based phylogenetic tree together with the current vaccine virus A/Michigan/45/2015. This suggests that the efficacy of the current seasonal flu vaccine could be about 50%, which corresponds to common protection against A(H1N1)pdm09 viruses in the last years. The problem could arise if viruses which are not compatible with the vaccine take over during the flu season. Of great concern are viruses with very high amplitude ratio A(0.29536)/A(0.055) that also carry pandemic markers (A/New Mexico/25/2018, A/California/70/2018, A/Texas/134/2018). In our opinion these viruses have increased pandemic potential and could represent precursors of the new pandemic virus.
In conclusion, presented results show: (i) that some pre-seasonal and seasonal A(H1N1)pdm09 viruses collected in October and November 2018 in the USA carry pandemic markers, indicating possibility of their evolution toward new pandemic viruses; (ii) that these US viruses have changed informational properties which determine their interacting profiles with the host, and; (iii) that about 50% of circulating viruses could escape the current flu vaccine and could evolve toward new pandemic viruses. Taking into account presented results, as well as the fact that A(H1N1)pdm09 viruses are dominant in the flu season 2018/2019, causing already high hospitalization and mortality rates in children and young healthy people, further monitoring of evolution of these viruses is urgently needed.
Sequence data of the viruses were obtained from the GISAID EpiFlu™ Database. To access the database each individual user should complete the “Registration Form For Individual Users”, which is available alongside detailed instructions. After submission of the Registration form, the user will receive a password. There are not any other restrictions for the access to GISAID. Conditions of access to, and use of, the GISAID EpiFlu™ Database and Data are defined by the Terms of Use.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)