Keywords
Genome graph, genome assembly
This article is included in the Python collection.
Genome graph, genome assembly
Sequence graphs are the core representation of genome assemblers1–3 Their use has increased lately thanks to the graphical fragment assembly (GFA) format for graph exchange4, tools to work with genome variation graphs5, and sequence to graph mappers6–10 But a lack of inter operation between graph-based tools, and limited tools for downstream graph-based analysis, contribute to a perceived complexity which maintains linear sequences as the typical unit of exchange. This flattening of graph representations within pipelines with multiple steps, that use different types of sequencing in an iterative fashion, produces ever-longer linear genome sequences through an information loss process. As a result, genome assembly projects are prone to error propagation and difficult to reproduce and control. These problems can be addressed developing graph-based frameworks to integrate the analysis of hybrid datasets.
The Sequence Distance Graph (SDG) framework implements a SequenceDistanceGraph representation that defines sequences in nodes and their adjacency in links, and an associated Workspace containing raw data and mappings. This provides an integrated working environment to use multiple sources of information to navigate and analyse genome graphs. Datastores allow random access to short, linked, and long read sequences on disk. A mapper on each datastore contains methods to map the reads to the graph and access the mapping data. KmerCounters provide functions to compute k-mer coverage over the graph from sequencing data, enabling coverage analyses. Additional DistanceGraphs, typically representing longer-range information and different linkage levels, define alternative topologies over the SequenceDistanceGraph nodes. Finally, a NodeView abstraction provides a proxy to a node, with methods to navigate the graph and access its mapped data. This comprehensive framework can be used to explore genome graphs interactively or to create processing methods for assembly or downstream analysis.
Here we describe the SDG implementation and basic tools, providing examples of use cases that highlight its analytic flexibility. First, we show how to create a hybrid assembly by integration of long reads linkage into a short-read graph. Then we analyse a simulated parent-child trio and show how the coverage of the parent datasets can be used to navigate the graph topology. These are only two of the multiple ways integrating data and genome graphs can be used to perform simple but powerful analyses.
The C++ core library implements SDG’s data structures and methods for WorkSpaces, graphs, datastores and mappers. Its main goal is to provide a straightforward interface to project information from raw datasets onto graphs, and enable easy access and analysis of the graph-data combination. It uses OpenMP for parallel processing, and SWIG 4.0 to export a Python API to enable interactive data analysis.
The SequenceDistanceGraph class contains a vector of nodes defining DNA sequences, and a vector of links. Every node has a positive and a negative end, and links are defined between these node ends. Links with positive distances represent gaps between linked sequences and negative distances represent overlaps. This representation, shown in Figure 1, is similar to those presented in 2,11 but unifies the concept of overlap and gap. Paths can be defined as list of nodes, with the sign of the first end in the walk. Graphs can be read and written to GFA and GFA2 files.
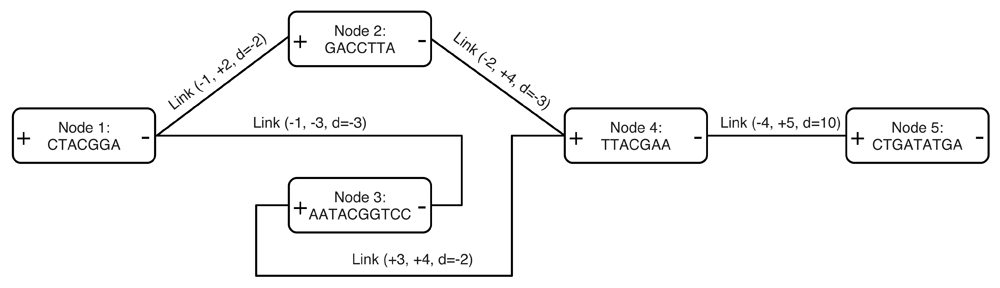
Sequences appear in only one direction and their reverse complement can be obtained by traversing the node in opposite direction, from - to +. The two largest possible paths are [1, 2, 4, 5] and [1, -3, 4, 5], and their reverse complements [-5, -4, -2, -1] and [-5, -4, 3, -1] respectively.
The DistanceGraph class contains a set of links over the nodes of a SequenceDistanceGraph object. It is used to represent alternative sources of linkage information, such as longer range linkage produced by mapped reads for scaffolding.
The WorkSpace contains a single SequenceDistanceGraph, multiple DistanceGraphs, datastores and mappers, and its structure in memory represents the status of the SDG framework. It can be dumped and loaded from disk, providing persistence and checkpoints between different steps on SDG-based pipelines. Raw reads and k-mer counts are kept in separate files, pointed from the WorkSpace, to avoid duplication when using multiple WorkSpaces around the same dataset.
The DataStores and Mappers provide access and management to raw data and its mapping on the graph. Datastores do not load read data into memory, but rather provide random access to the on-disk data. The PairedRedMapper and LinkedReadMapper classes use a unique k-mer index to map reads to single nodes, with single reads mapping to multiple nodes not being mapped3,12 The LongReadMapper class generates multiple mappings from each read to nodes, using a short non-unique k-mer index (k=15 by default)13,14 Long read mapping filtering is left to later stages of the processing.
The KmerCounters creates an index with all the k-mers at a given k up to k=31 and counts occurrences of these k-mers on the graph, allowing then to count occurrences in datastores or fastq files. These counts, persisted in the KmerCounter with a name, can be then accessed to perform k-mer coverage analyses. Projections of raw k-mer coverage in the reads and the assembly over a particular sequence for a node or path, similar to those produce by the "sect" tool of K-mer Analysis Toolkit (KAT)15 are valuable for content analysis. Spectra analysis of these frequencies can provide further insight into genome composition and representation on the assembly.
Two processing classes, LinkageUntangler and LinkageMaker, work with alternative linkage configurations. The LinkageMaker is used to condense information via one of its make_linkage* methods, from evidence in the WorkSpace into links in a DistanceGraph. The LinkageUntangler class works on a DistanceGraph to simplify, condense and/or linearise its linkage. In the second use case below it can be seen how a combination of LinkageMaker and LinkageUntangler can be used for scaffolding with long reads.
Finally, the NodeView class, and its associated LinkViews, provide a single-entry point for node-centric analyses. A NodeView from either a DistanceGraph or SequenceDistanceGraph is a wrapper containing a pointer to the graph and a node id, and will provide access to its nodes’ previous and next linked nodes, mapped reads, or k-mer coverage. A user with good understanding of the NodeView class should be able to access most information in the WorkSpace through it, making it the default choice for analysing the graph.
Requirements and installation. SDG can be run on Linux and MacOS, and requires enough RAM to hold the WorkSpace completely in memory, which will depend on the dataset. Space to hold the uncompressed sequences on the datastores on disk will also be required.
SDG can be installed via pre-compiled binaries from https://github.com/bioinfologics/sdg/releases. The binaries have been built using Python3 and GCC version 6 from the Ubuntu package manager for the Linux version. The MacOS version dependencies were obtained using Homebrew (Python3, GCC-6 and SWIG). SDG can be compiled using CMake, Python3, SWIG version 4 and GCC version 6 onwards. Detailed instructions can be found at https://bioinfologics.github.io/sdg/sdg/README.html#installation.
Typical workflow. Working with SDG typically involves two different stages: creating a WorkSpace with the data and mappings, and analysing this WorkSpace. SDG includes command line tools to create DataStores, KmerCounts, and WorkSpaces, and map reads within a WorkSpace.
sdg-datastore: creates a Datastore from raw reads and can process paired, 10x or long reads. An output prefix is specified as a parameter and a <prefix>.prseq, <prefix>.lrseq or <prefix>.loseq file is generated.
sdg-kmercounter: creates a KmerCounter indexing a graph from a WorkSpace or GFA, or works with an already generated one. A count can be added directly from raw reads or from a datastore. The KmerCounter is persisted on file with extension ’sdgkc’.
sdg-workspace: creates a WorkSpace from a base graph or works with an already generated one. Datastores and KmerCounters can be added. The WorkSpace is persisted on file with extension ’sdgws’.
sdg-dbg: creates a WorkSpace from a PairedReadDatastore by building a deBruijn graph and using this as the base graph. Counts for the k-mers from the graph and raw reads are added too.
sdg-mapper: maps reads within a WorkSpace. An updated WorkSpace is produced and dumped to the specified prefix.
WorkSpaces can also be instantiated with an empty graph, and the graph populated through the add_node and add_link methods. The following example on a python session shows how the simple graph from Figure 1 can be created from scratch, navigated through a NodeView instance and sequence from its paths extracted.
>>> import pysdg as SDG version 0.1 master b4d3f02 >>> ws=SDG.WorkSpace() >>> ws.sdg.add_node("CTACGGA") 1 >>> ws.sdg.add_node("GACCTTA") 2 >>> ws.sdg.add_node("AATACGGTCC") 3 >>> ws.sdg.add_node("TTACGAA") 4 >>> ws.sdg.add_node("CTGATATGA") 5 >>> ws.sdg.add_link(-1, 2, -2) >>> ws.sdg.add_link(-1, -3, -3) >>> ws.sdg.add_link(-2, 4, -3) >>> ws.sdg.add_link(3, 4, -2) >>> ws.sdg.add_link(-4, 5, 10) >>> nv=ws.sdg.get_nodeview(1) >>> nv <NodeView: Node 1 in SDG> >>> nv.next() <Vector: 2 LinkViews> >>> print(nv.next()) [ <LinkView: -3bp to Node -3>, <LinkView: -2bp to Node 2> ] >>> nv = nv.next()[0].node() >>> nv <NodeView: Node -3 in SDG> >>> print(nv.prev()) [ <LinkView: -3bp to Node 1> ] >>> nv.sequence() 'GGACCGTATT' >>> SDG.SequenceDistanceGraphPath(ws.sdg, [1, -3, 4, 5]).sequence() 'CTACGGACCGTATTACGAANNNNNNNNNNCTGATATGA'
Typically, as shown in Figure 2, the API is used to explore a larger WorkSpace, with the methods accessing both in-memory and on-disk data, and modifying the status of the WorkSpace.
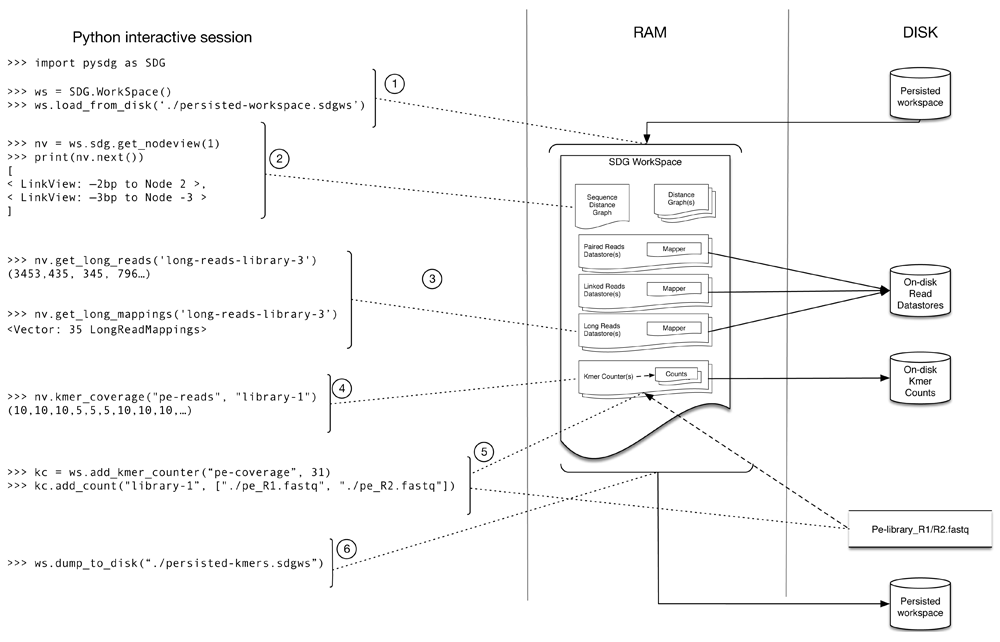
The WorkSpace holds the information for a project and contains the graphs, the mappers and k-mer counts. From Python, a previously saved WorkSpace is loaded from disk (1). The NodeView object is centred on a specific node and can be used to access node characteristics (ie. size and sequence), graph topology from the perspective of the node you are on (i.e. neighbours in both directions (2)) and can also retrieve information projected onto the selected node (ie. mappings (3) and k-mer coverage (4)). Operations such as adding a KmerCounter to the WorkSpace and adding a count (5) can be performed, and the WorkSpace can be saved back to disk (6). Once loaded, the bulk of the WorkSpace is held in memory for fast access with the raw read data from the DataStores remaining on disk accessible through random access.
To illustrate the use of SDG, we have reproduced a short version of two examples from http://bioinfologics. github.io/sdg_examples.
All paired end datasets are available on https://zenodo.org/record/3363871#.XUwyVy2ZN2416, and the PacBio reads are from NCBI accession PRJNA19443717 For simplicity, we have also made the datasets available on https://opendata.earlham.ac.uk/opendata/data/sdg_datasets/ as ready-to-use ’fastq.gz’ files.
This example is based on an E. coli dataset combining PacBio reads from 17 and Illumina Miseq 2x300bp reads subsampled from a test run. It uses the long reads to scaffold a short read based graph produced by sdg-dbg. Graphs are dumped to GFA files at different stages, and visualised using Bandage v0.8.118
First, we use the command line tools to create datastores for both long and short reads and an initial WorkSpace containing a DBG assembly:
sdg-datastore make -t paired -o ecoli_pe ../ecoli_pe_r1.fastq.gz -2 ../ecoli_pe_r2.fastq.gz sdg-datastore make -t long -o ecoli_pb -L ../ecoli_pb_all.fastq.gz sdg-dbg -p ecoli_pe.prseq -o ecoli_assm
From this point on, we use the python SDG library. First, we load the workspace, add a long read datastore and map its reads using a k=11 index.
import pysdg as SDG # Load sdg-dbg's workspace from disk, add the pacbio datastore ws = SDG.WorkSpace('ecoli_assm.sdgws') lords = ws.add_long_reads_datastore('ecoli_pb.loseq') # Map long reads lords.mapper.k = 11 lords.mapper.map_reads() ws.sdg.write_to_gfa1('initial_graph.gfa')
The graph, as shown in Figure 3A contains multiple unresolved repeats.

Linkage at different stages of the long read scaffolding example, visualised using Bandage: A) SequenceDistanceGraph generated by sdg-dbg from short reads, B) DistanceGraph generated after using make_nextselected_linkage on the long read data, linking all nodes of 1100bp and more, C) DistanceGraph after eliminating all nodes with multiple connections (repeats).
We can use the LinkageMaker to create linkage using the long reads datastore. We do this by selecting the nodes between which to analyse possible linkage, in this case all nodes of 1100bp or more, and then calling the make_longreads_multilinkage method, with alignment filtering parameters of 1000bp and 10% id.
lm = SDG.LinkageMaker(ws.sdg) lm.select_by_size(1100) mldg = lm.make_longreads_multilinkage(ws.long_reads_datastores[0].mapper, 1000, 10)
This multi-linkage can be collapsed using the LinkageUntangler. The make_nextselected_linkage method links every selected node to its closest selected neighbours on each direction, aggregating the distances via a simple median calculation:
lu = SDG.LinkageUntangler(mldg) lu.select_by_size(1100) ns_dg = lu.make_nextselected_linkage() ns_dg.write_to_gfa1('ns_collapsed.gfa')
The new graph we dumped, as shown in Figure 3B, has disconnected the repeats and introduced long read linkage which skips over them, but it is still not fully solved. We can improve this further by getting rid of repetitive nodes that will be connected to multiple neighbours, as each of them belongs in more than one place. We do that by just turning these nodes’ selection off in the LinkageUntangler, which will then skip them in the solution.
for nv in ns_dg.get_all_nodeviews(): if len(nv.prev()) > 1 or len(nv.next()) > 1: lu.selected_nodes[nv.node_id()] = False ns_nr_dg = lu.make_nextselected_linkage() ns_nr_dg.write_to_gfa1('ns_nr_final.gfa')
The last graph is now a circle, with all the repeats disconnected from any linkage.
We created a simulation of a trio dataset for this example using the synthetic genome creation and sequencing package Pseudoseq.jl v0.1.019 Chromosomes 4 and 5 of the reference genome of the yeast strain S288C were used as templates to create a diploid, genome for each parent with 1% heterozygous sites. Each homologous pair of chromosomes was crossed over and recombined and the child inherited one chromosome from the first parent at random, and one chromosome from the second parent at random. Simulated paired end reads were generated for each genome, using an average fragment length of 700bp and a read length of 250bp, and an expected coverage of 70x with error rate was set to 0.1%.
First we used the command line tools to create a graph from the child reads using sdg-dbg, and add k-mer counts for both parents into the datastore.
sdg-datastore make -t paired -1 child/child-pe-reads_R1.fastq.gz -2 child/child-pe-reads_R2.fastq.gz -o child_pe
sdg-dbg sdg-dbg -p child_pe.prseq -o sdg_child
sdg-kmercounter add -c main.sdgkc -n p1 -f p1/p1-pe-reads_R1.fastq.gz -f p1/p1-pe-reads_R2.fastq.gz -o main
sdg-kmercounter add -c main.sdgkc -n p2 -f p2/p2-pe-reads_R1.fastq.gz -f p2/p2-pe-reads_R2.fastq.gz -o mainWe now open the WorkSpace and use the NodeView::parallels method to look for the largest bubble structure in the graph, which should be formed by two parallel nodes with haplotypes coming from each parent.
import pysdg as SDG ws = SDG.WorkSpace('sdg_child.sdgws') #Largest node with one parallel node, and its parallel maxbubble = 0 for nv in ws.sdg.get_all_nodeviews(): if nv.size() > maxbubble and len(nv.parallels()) == 1: maxbubble=nv.size() bubble_nvs=(nv,nv.parallels()[0])
Since each side should be a haplotype from a different parent, we should see a loss of k-mer coverage on the parent that didn’t contribute that haplotype. To check this, we create a plotting function to plot the output from the NodeView::kmer_coverage method.
def plot_kcov(nv): '''Plot kmer coverage across the three read sets. Requires pylab.''' figure();suptitle("Coverage for "+str(nv)); subplot(3,1,1);ylim((0,120)) plot(nv.kmer_coverage("main","PE"), label="child"); legend(loc=1); subplot(3, 1, 2);ylim((0, 120)) plot(nv.kmer_coverage("main","p1"), "red", label="parent 1"); legend(loc=1); subplot(3, 1, 3);ylim((0, 120)) plot(nv.kmer_coverage("main","p2"),"blue", label="parent 2"); legend(loc=1); plot_kcov(bubble_nvs[0]) plot_kcov(bubble_nvs[1])
The plots, shown in Figure 4, reflect how Node 4775 contains content inherited from parent 2 and its parallel node 11414 contains content inherited from parent 1. We can create a function to extend these parent-specific regions by walking forward and backward as long as only one link takes us to a node that is fully covered by the content of the parent we are following.
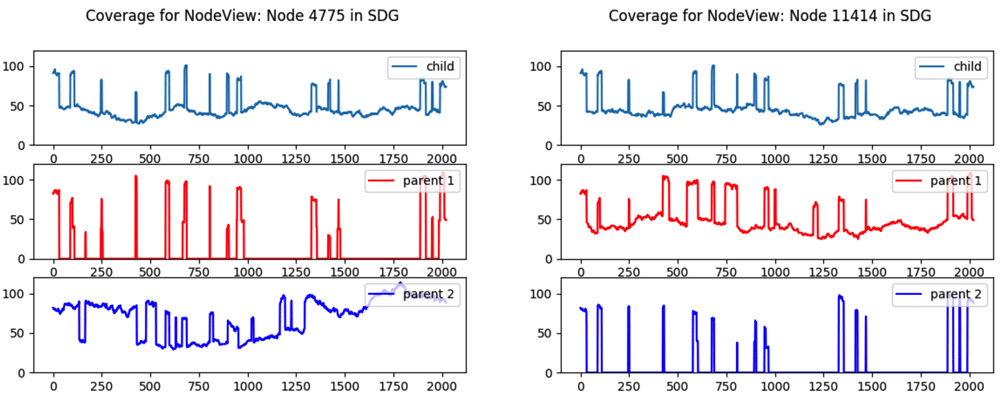
Coverage drops to 0 on the opposite parent for k-mers that are unique to a parent.
def extend_parent_covered_path(starting_node,target_parent): if ws.sdg.get_nodeview(starting_node).kmer_coverage("main", target_parent).count(0) != 0: return SDG.SequenceDistanceGraphPath(ws.sdg,[]) p = SDG.SequenceDistanceGraphPath(ws.sdg,[starting_node]) for x in [0,1]: nv = ws.sdg.get_nodeview(p.nodes[-1]) while nv.next(): next_node = 0 for nl in nv.next(): if nl.node().kmer_coverage("main", target_parent).count(0) == 0: if next_node or nl.node().node_id() in p.nodes: next_node = 0 break else: next_node = nl.node().node_id() if next_node == 0: break p.nodes.append(next_node) nv=ws.sdg.get_nodeview(next_node) p.reverse() return p path1=extend_parent_covered_path(11414, "p1") path2=extend_parent_covered_path(4775, "p2")
After using this function, path1 contains 49 nodes yielding 8672bp of sequence inherited from parent 1, and path2 contains 139 nodes yielding 26351bp of sequence inherited from parent 2. It is important to note that the difference in node count and sequence length arises because the extension function is haplotype-specific and its results depend in the topology of each haplotype graph.
The Sequence Distance Graph framework provides a unified workspace for different sequencing technologies using the genome graph as the basis of integration. It enables analyses across the graph topology, the raw data and its projections to the graph. We have shown how the NodeView class can be used through the Python API to produce interactive analyses that are both powerful and easy to follow. We expect this will be a useful codebase for all levels of users, not only for the construction of graph-based analysis but also for their teaching and dissemination.
The PacBio, E. coli reads are deposited on NCBI accession PRJNA194437 from Koren et al.17
E. coli K12 Re-sequencing with PacBio RS and 454: Accession number PRJNA194437, https://identifiers.org/ncbi/bioproject:PRJNA194437
The datasets used in the examples are available from: https://opendata.earlham.ac.uk/opendata/data/sdg_datasets/ and archived in Zenodo Zenodo: SDG Paper Datasets. http://doi.org/10.5281/zenodo.336387116
Data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
Software documentation: https://bioinfologics.github.io/sdg
Source code available from: http://github.com/bioinfologics/sdg
Archieved source code at time of publication: https://zenodo.org/record/3363165#.XUw1yy2ZN2520
License: MIT License
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)