Keywords
Hong Kong Chinese, LPIN1, Rhabdomyolysis, Novel variants
Hong Kong Chinese, LPIN1, Rhabdomyolysis, Novel variants
Rhabdomyolysis is an uncommon but potentially fatal condition. It results from an acute muscle fiber breakdown resulting in the release of intracellular muscle constituents into the circulation. There are multiple causes for rhabdomyolysis. However, recurrent rhabdomyolysis is frequently attributed to metabolic myopathies. The differential diagnoses include fatty acid ß-oxidation defects (FAOD), mitochondrial respiratory chain disorders and glycogen storage diseases. When initial investigations including biochemical metabolic work-up are unremarkable, muscle biopsies for histological and enzymatic studies are commonly performed. In recent years, autosomal recessive mutations in LPIN1 are being increasingly recognized as an important cause of acute recurrent rhabdomyolysis in childhood in the Western population, after the exclusion of FAOD, mitochondrial respiratory chain disorders and glycogen storage diseases. Lipin 1 (LPIN1), encoded by LPIN1 gene, belongs to the LPIN gene family which also include LPIN2 and LPIN3. Lipin 1 is predominantly expressed in skeletal muscle and adipose tissue1–3. Among lipins 1, 2 and 3, lipin 1 is the only lipin which is expressed significantly in muscles2,4. Lipin 1 is a Mg2+-dependent phosphatidic acid phosphohydrolase (PAP) that catalyzes the dephosphorylation of phosphatidic acid to yield diacylglycerol and inorganic phosphate5. It has also been found to exert transcriptional co-regulator activity6,7. The human LPIN1 gene is mapped to chromosome 2p25.1 and contains 20 exons. Apart from acute recurrent rhabdomyolysis, LPIN1 variants have also been found in adults with statin-induced myopathy, metabolic syndrome and type 2 diabetes mellitus8–11. We hereby report the first probable Hong Kong Chinese case of acute recurrent rhabdomyolysis in a boy with compound heterozygous LPIN1 variants who presented with recurrent rhabdomyolysis since the age of 15 months.
A 15-month-old boy was brought to the hospital complaining of coryzal symptoms for three days, and dyspnea and fever for one day. He was seen by a general practitioner a day prior to the admission and was given an intramuscular injection of an uncertain drug for his illness. After the injection, he complained of passing dark-colored urine twice. Physical examination showed good perfusion and the patient’s blood pressure was 113/65 mmHg. His body weight and height were at the 75th percentile and 50th percentile, respectively, for his age and sex. Respiratory examination revealed bilateral wheezes and the initial impression was acute bronchiolitis. Creatine kinase (CK) was markedly elevated to 127,494 U/L (reference interval: 39–308 U/L). Urine dipstick was positive for hemoglobin. After centrifugation of the urine through a 30 kDa microconcentrator membrane, urine dipstick for hemoglobin remained positive, confirming that the positive urine dipstick result without ultrafiltration was likely due to the presence of myoglobin, which cross-reacts with the peroxidase reaction employed by the urine dipstick for hemoglobin testing. Plasma creatinine and potassium were normal. Liver function tests revealed an elevated alanine aminotransferase (ALT) of 658 U/L (reference interval: 9–25 U/L) on the first day of admission; the value peaked on the second day of admission with a reading of 1,418 U/L. Apart from CK, lactate dehydrogenase (LDH) level was also raised to 9,154 U/L (reference interval: <350 U/L) and it peaked on the second day of admission at a value of 11,409 U/L. Cardiac troponin-I level was normal and electrocardiogram (ECG) did not show any abnormalities. A diagnosis of rhabdomyolysis was made.
Intravenous fluid was given in view of poor appetite and later tapered off as diet was better tolerated. CK came down to 10,909 U/L on the fourth day of admission. The boy had good urine output and there was no more passage of dark-colored urine since day three of the admission. Nasopharyngeal swab taken during admission was positive for respiratory syncytial virus. Dyspnea gradually resolved. On day five of illness, the CK, ALT and LDH had markedly improved. Repeated urine myoglobin and hemoglobin testing were negative. He was followed up two weeks later and was well then. There was no more passage of dark-colored urine.
Thereafter, he defaulted follow up and presented to us again at six years of age. This time, he complained of bilateral calf pain with antalgic gait associated with coryzal symptoms and fever. The episode was not preceded by any injuries nor excessive exercises. There was also no preceding administration of any intramuscular injection. Upon detailed history taking, the patient revealed that he frequently got muscle cramps after exercise. There was no family history of recurrent rhabdomyolysis, malignant hyperthermia or musculoskeletal diseases. The parents were both Chinese and non-consanguineous.
Physical examination revealed bilateral calf tenderness in the absence of weakness. There was no associated hepatosplenomegaly. He was well built with a weight and height at the 50th and 75th percentile, respectively, for his age and sex. Investigations during this admission again revealed an elevated CK (27,442 U/L), LDH (658 U/L) and ALT (138 U/L). Urine for myoglobin and hemoglobin (analyzed by same methods as before) were both positive. Plasma creatinine and potassium were normal. Hydration was instituted and the CK levels normalized 10 days later. Due to the recurrent episodes of rhabdomyolysis, further investigations were undertaken including blood gas, plasma lactate and ammonia but they were all unremarkable. Metabolic screening was performed and showed a normal pattern of plasma amino acids, serum acylcarnitine profile and urine organic acid profile. Genetic analysis of all coding exons of LPIN1 reviewed two heterozygous novel mutations: NM_145693.2(LPIN1):c.[1949_1967dupGTGTCACCACGCAGTACCA]; [2410G>C] (p.[Gly657Cysfs*12];[Asp804His]) (Figure 1). The parents also underwent genetic tests for LPIN1. Each of the parents was a heterozygous carrier of one of the patient’s variants, which confirmed compound heterozygosity of the variants in the patient. The former variant was classified as likely pathogenic while the latter variant was classified as a variant of uncertain significance (VUS). Although the genetic findings were inconclusive, the patient’s clinical presentation was compatible with LPIN1-related acute recurrent rhabdomyolysis, and muscle biopsy was not done.
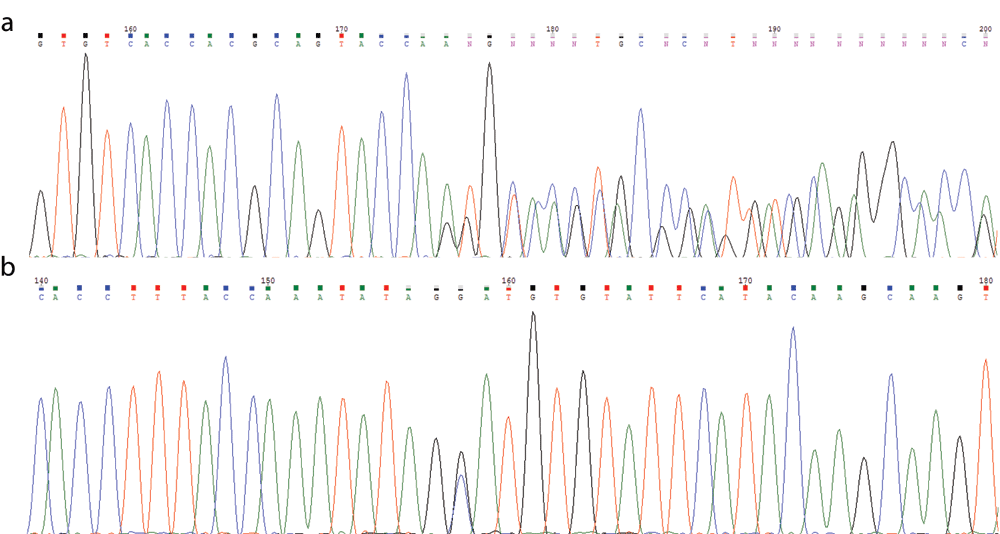
(a) NM_145693.2(LPIN1):c.[1949_1967dupGTGTCACCACGCAGTACCA] p.[Gly657Cysfs*12]. (b) NM_145693.2(LPIN1):c.[2410G>C] p.[Asp804His].
In spite of the uncertainty in the genetic findings, the likely diagnosis of LPIN-related acute recurrent rhabdomyolysis was conveyed to the family with proper genetic counseling conducted. Advice to avoid recurrent episodes of rhabdomyolysis, which include limiting strenuous exercise, ample hydration during exercise and prompt medical consultation in case of myalgia, was given. One year after the genetic diagnosis, he has been growing well and did not complain of passing any dark-colored urine, even during febrile illness.
The parents decided to have another baby and opted not for prenatal testing for LPIN1 variants. Prenatal testing carries intrinsic risk to the fetus and, in our opinion, is not recommended for this disease which is amendable to treatment and not debilitating. Post-natal genetic test on the younger sister confirmed that she also carried the two familial heterozygous variants; upon this finding, counselling was given to the parents.
LPIN1 mutations resulting in recurrent rhabdomyolysis was first described in three patients by Zeharia et al. in 200812. In less than a decade since it was first described, there has been an increasing evidence in the literature that LPIN1 mutations are one of the most frequent causes of recurrent rhabdomyolysis in childhood. Michot et al. reported that after excluding primary fatty acid oxidation disorders, recessive LPIN1 gene mutations were found in 59% of the patients who exhibited severe recurrent rhabdomyolysis in childhood. The male to female ratio was reported to be 0.89. Out of the 17 cases described by Michot et al., more than half of them were Caucasian and the others were of African, Asian (Vietnamese) and Maghrebi ethnicity13. Due to its relative novelty, the prevalence of the disease is still unknown. To our knowledge, there has been no previous report in the English-language literature of a child with Chinese ethnicity and LPIN1-related acute recurrent rhabdomyolysis (MIM #268200).
The pathophysiology of LPIN1-related recurrent rhabdomyolysis is not fully understood. Accumulation of lipid droplets is a common finding in muscle biopsy14. The loss of PAP enzymatic activity, rather than a loss in transcription co-regulator activity, plays a major role in the pathogenesis of this disease as the loss of the PAP activity alone without a loss in transcription co-regulator activity was sufficient to cause rhabodomyolysis15. Apart from the PAP activity responsible for triacylglycerol and phospholipid biosynthesis, it also serves as a transcriptional co-activator together with peroxisome proliferator activated receptor coactivator-1α (PPARA) and peroxisome proliferator activated receptor gamma coactivator-1 alpha (PGC-1α) to regulate genes encoding fatty acid oxidation and respiratory chain enzymes6,7. Multiple genes are either up-regulated or down-regulated in myoblasts of lipin-1-deficient patients16. Furthermore, various inflammatory inducers including lipopolysaccharides, zymosan and proinflammatory cytokines have been shown to repress LPIN1 expression in adipose tissue and muscular tissue17,18. Inflammatory inducers are usually produced when the body is under catabolic stress and the resulting suppressed lipin 1 activity may lead to rhabdomyolysis. This hypothesis might explain the fact that the majority of the severe episodes are associated with catabolic states such as a preceding illness13. Lipin 1 was also found to play in a role in autophagy of mitochondria as PAP activity is required for the maturation of autolysosomes and lipin 1 deficiency leads to accumulation of aberrant mitochondria19. This may explain the finding of mitochondrial aggregates on ultrastructural examination of muscle tissue in some patients14. Sarcoplasmic reticulum stress response was recently identified to be an important player in the pathogenesis of this disease by contributing to catastrophic de novo lipogenesis of both phospholipids and neutral lipids20.
Disease-causing LPIN1 mutations are scattered throughout the coding region14. An intragenic deletion (c.2295-863_2410-27del) is frequently seen in Caucasian patients, possibly representing a founder effect13,14. In our patient, two heterozygous novel variants of LPIN1 gene are detected: the 19-nucleotide duplication NM_145693.2(LPIN1):c.[1949_1967dupGTGTCACCACGCAGTACCA] (p.[Gly657Cysfs*12]) causes a frameshift with a premature termination codon and hence has deleterious effect; the novel missense variant, NM_145693.2(LPIN1):c.[2410G>C] (p.[Asp804His]), is found in the highly conserved LNS2 (Lipin/Ned1/Smp2) domain near the C terminus of lipin 1, which contains an important Mg2+-dependent catalytic site responsible for the PAP activity21. This amino acid residue is highly conserved. The variant is predicted to be deleterious (SIFT), damaging (PROVEAN) and probably damaging (Polyphen-2) by in silico analyses. At the time of reporting, both variants are absent from controls in the Exome Sequencing Project, 1000 Genomes Project, Exome Aggregation Consortium and Genome Aggregation databases. NM_145693.2(LPIN1):c.[1949_1967dupGTGTCACCACGCAGTACCA] (p.[Gly657Cysfs*12]) was classified as a likely pathogenic variant (Criteria: PVS1 and PM2) and NM_145693.2(LPIN1):c.[2410G>C] (p.[Asp804His]) was classified as a variant of uncertain significance (VUS)(Criteria: PM2, PM3 and PP3) based on the American College of Medical Genetics and Genomics (ACMG) guideline published in 201522. Functional characterization of the two detected variants is warranted to confirm their pathogenicity.
All patients with LPIN1 mutations present with recurrent rhabdomyolysis triggered by febrile illness, prolonged fasting or anesthesia12,13. Michot et al. reviewed 29 cases with recurrent rhabdomyolysis, in which 17 patients were confirmed to harbor LPIN1 mutations. For the patients who harbor LPIN1 mutations, the first episodes of rhabdomyolysis occurred before the age of five years. The mean age of presentation was 21 months old and the earliest age of presentation was at five months of age. The number of recurrent episodes ranged from one to ten per patient13. Our patient presented at 15 months of age and has so far experienced two episodes of rhabdomyolysis. As more than 50% of patients with recurrent rhabdomyolysis were found to have LPIN1 mutations after exclusion of FAOD13, genetic testing for LPIN1 mutations should be performed prior to muscle biopsy if metabolic investigations are unremarkable. Muscle biopsy is an invasive procedure and require general anesthesia which may trigger rhabdomyolysis.
Michot et al. also noted that among the 29 cases of unexplained rhabdomyolysis they studied, all cases eventually confirmed to have LPIN1 mutations had a CK level of >10,000 U/L13. In our patient, the CK levels were 127,494 U/L and 27,442 U/L during the first and second episodes of rhabdomyolysis, respectively. This is in line with Michot’s findings. Therefore, one should be vigilant to look for LPIN1 mutations in any young children presenting with rhabdomyolysis with a CK level of >10,000 U/L.
The management of rhabdomyolysis is mostly supportive by providing aggressive fluid replacement therapy to protect the renal function. Besides, close monitoring is also indispensable during fasting and anesthesia. As rhabdomyolysis episodes could be fatal, prevention of such events is of utmost importance. Parents should be well educated on the symptoms and precipitating factors of rhabdomyolysis. They should also be given an action plan when the child exhibits muscle pain or fever. In our case, proper education of the parents at diagnosis has helped in preventing further episodes of rhabdomyolysis. In a group of five patients with biallelic LPIN mutations, Pichler et al. proposed that high-caloric intake at periods of stress and intravenous glucose during episodes of rhabdomyolysis may decrease the number of rhabdomyolysis episodes and the duration of each episode by preventing catabolism22. Further studies are required to confirm the efficacy and safety of this approach.
For LPIN1-related acute recurrent rhabdomyolysis, it was reported that mortality was as high as 30% during an acute episode of severe rhabdomyolysis14. Between episodes, these patients thrived well. Intellectual disability was reported in one case at the age of nine years12. Our patient has been normal between the episodes apart from frequent muscle cramps after exercise and there is no evidence of intellectual disability. However, further observation is needed in view of the young age of our patient. The long-term prognosis of this condition has been reported in a 25-year-old female with LPIN1-related recurrent rhabdomyolysis, who had bilateral common peroneal neuropathies in addition to a background residual distal myopathy detected one year following discharge from intensive care. It was uncertain whether the neuropathies were a result of critical illness, compression and/or severe weight loss during the admission or intrinsic to the underlying genetic lipin 1 deficiency23.
Genetic confirmation of LPIN1 mutations not only helps the patient but also their family members. In our case, the younger sister had genetic testing done soon after birth. This has raised the parents’ awareness of the disease and its preventive measures. The younger sister has been growing well and has had no episodes of rhabdomyolysis at the time of reporting at one year of age.
This is the first probable reported case of LPIN1-related rhabdomyolysis in the Hong Kong Chinese population. It has been shown that LPIN1-related rhabdomyolysis is a major cause of severe rhabdomyolysis in early childhood so we should be vigilant of this condition. Screening for LPIN1 mutations should be undertaken at an early stage in the work up of childhood rhabdomyolysis. As rhabdomyolysis in children has a high mortality rate, early recognition of the symptoms, timely diagnosis and proactive management are essential. Regarding prognosis, more observations and longitudinal studies are warranted to determine the long-term outcomes of this condition.
NCBI ClinVar: NM_145693.2(LPIN1):c.[1949_1967dupGTGTCACCACGCAGTACCA] (p.[Gly657Cysfs*12]). Accession number SCV000965694.
NCBI ClinVar: NM_145693.2(LPIN1):c.[2410G>C] (p.[Asp804His]). Accession number SCV000965695.
Written informed consent for publication of the clinical details was obtained from the parents of the patient.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)