Keywords
bioinformatics, genomics, genetics, GWAS, variant annotation, SNPs, software, clinical genetics, epigenetics, epigenomics
This article is included in the Research to the People collection.
bioinformatics, genomics, genetics, GWAS, variant annotation, SNPs, software, clinical genetics, epigenetics, epigenomics
Genome sequencing has become an invaluable tool for clinical diagnostics. Several methodologies exist to look at the human genome, each with own benefits and pitfalls. Whole exome sequencing has been a cost-effective technique to identify structural and nucleotide variants in protein-coding regions of the genome, and has identified variants associated with a number of diseases, including psoriasis1, Factor V Leiden thrombophilia2, and Miller Syndrome3. Genome-wide associations studies, originally based on SNP microarrays, have found that roughly half of genetic associations with disease are located outside gene bodies; this fraction approaches 90% with the inclusion of intronic regions4. With dropping costs for DNA sequencing, whole genome sequencing (WGS) promises to extend the ability to identify disease-associated single and structural nucleotide variants in the clinic, to non-coding regions of the genome, including gene and chromatin regulatory sequences. However, the increased size and complexity of the data creates a parallel challenge of annotating non-coding variants, as it requires knowledge of tissue-specific gene regulation (or epigenetics), including regulatory elements such as promoters and enhancers, and features of higher order chromatin organization such as Topological Associated Domains.
Several large-scale epigenomics projects have catalogued tissue-specific regulatory elements. This includes genetic modulation of gene expression (GTEx project)5, chromatin state (Roadmap Epigenomics)6, and enhancer-promoter loops for mapping of distal regulatory elements to genes (FANTOM and individual studies)7,8. A computational workflow that integrated these functional annotation maps to annotate variants from WGS assays would be a valuable resource to prioritize variants with potential functional impact in tissues of interest. Popular high-throughput variant annotation tools, such as BioMart and Variant Effect Predictor9,10, do not provide tissue-specific annotation. While FUMA11 integrates comprehensive epigenetic annotation, it is a web-based service used to annotate top-ranking variants from GWAS studies, rather than being a standalone tool for variant annotation.
In this work, we describe and provide variant annotation software that starts with output from a WGS assay and prioritizes variants based on epigenomic resources described above, as well as clinical genetic and GWAS catalogs of variant-disease association. This tool will allow users to capture functional and clinical information and to analyze variants simply by providing the commonly-used VCF file format. We demonstrate the software's functionality by prioritizing variants from WGS data for a single patient.
This work was undertaken as part of SVAI Undiagnosed-1 (https://sv.ai/undiagnosed-1), which was a collaborative event with the goal of diagnosing a patient with an unknown genetic condition. As data, participant groups were provided with detailed medical history, genotyping and metabolic data from a 33-year old Caucasian male patient, JCM. The event was hosted in June 2019 by the not-for-profit organization SVAI (http://sv.ai), with participants located in the San Francisco Bay Area (USA) as well as in Toronto, Canada.
Figure 1 shows the workflow for the pipeline. Patient genotypes are provided in Variant Call Format as input. Conceptually, the tool compiles prior knowledge about the functional significance of variants from the perspective of tissue-specific regulatory information, known genetic disease associations in the literature, tissue-specific genetic modulation of gene expression, and clinical pathogenicity. The annotation sources are integrated with the genotype calls in the VCF file, and this integration results in a single output table with available annotation for the variant. The user can then prioritize variants based on the combination of known or predicted functional consequences.
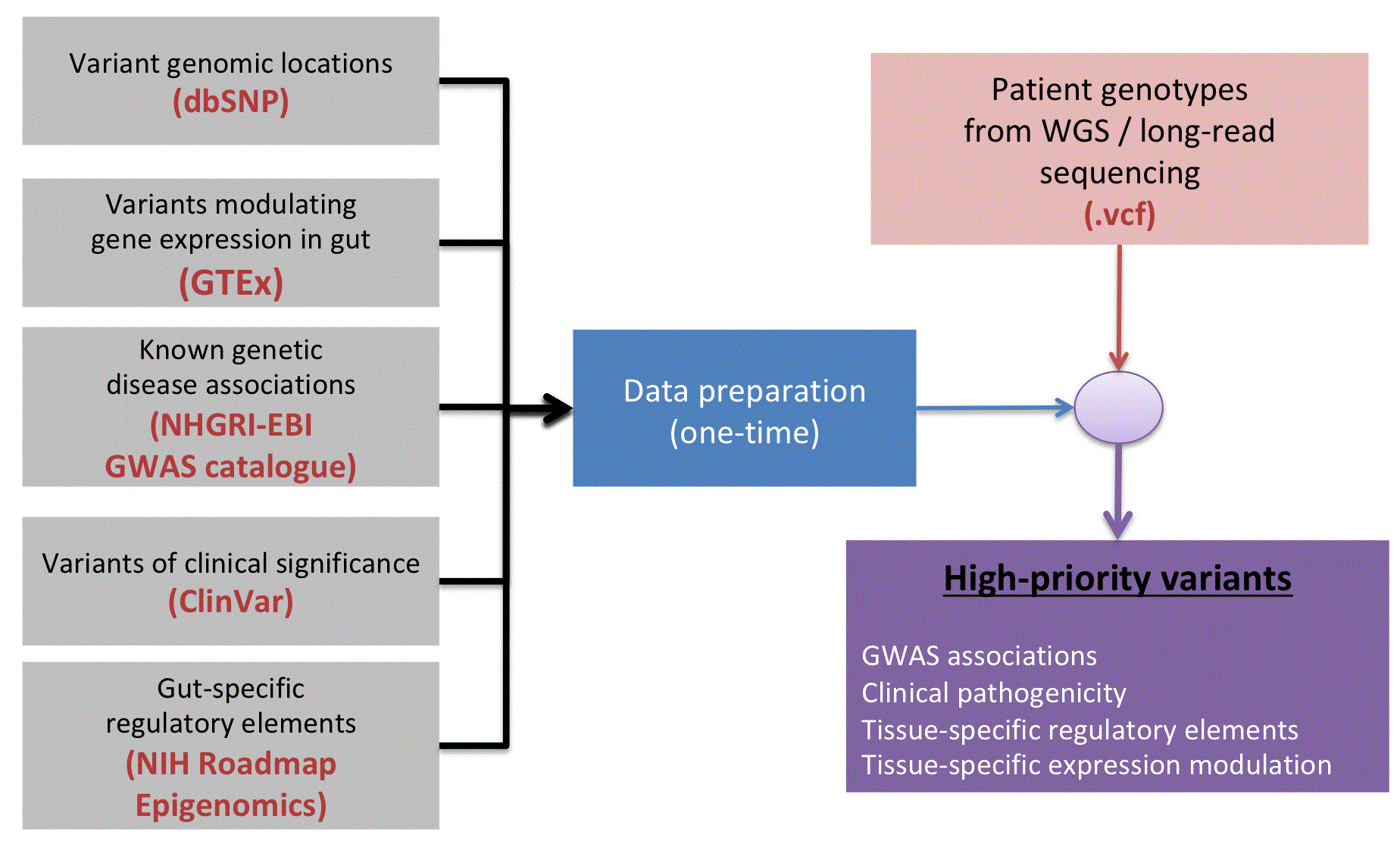
The NIH Roadmap Epigenomics project performed comprehensive mapping of noncoding DNA in 111 epigenomes to identify putative regulatory elements in diverse human tissue and cell types (http://www.roadmapepigenomics.org/)6. The presence of a variant overlapping a tissue-specific promoter or enhancer element signifies that variant alteration could change the regulation of a tissue-specific gene, i.e. that it has regulatory impact on gene expression. Tissue-specific 15-state chromatin state models were downloaded from the Roadmap Epigenomics Portal (downloaded from https://egg2.wustl.edu/roadmap/data/byFileType/chromhmmSegmentations/ChmmModels/coreMarks/jointModel/final/all.mnemonics.bedFiles.tgz). As the symptoms for John M, hereafter referred to as “the Patient”, included impaired function of the gastrointestinal tract, we limited our annotation to tissues and organs of the digestive system (e.g. esophagus, stomach, intestine). Chromatin states for digestive system tissues were used, including fetal stomach, small and large intestine, sigmoid colon, colonic mucosa, mucosa from stomach, duodenum and rectum, esophagus, rectal mucosa, and stomach mucosa. Regions with open chromatin were included for variant annotation (Table 1; states ≤ 1–7).
The GTEx project identified variants that significantly modulate gene expression in each of 44 human tissues5. Variants that modulate transcription in gut tissues (gut eQTLs) were downloaded from the GTEx portal (downloaded from https://storage.googleapis.com/gtex_analysis_v7/single_tissue_eqtl_data/GTEx_Analysis_v7_eQTL.tar.gz; Table 2).
Genome-wide SNP-disease associations were downloaded from the NHGRI-EBI GWAS catalog12, using the TargetValidation.org API13; only those associations mapped to metabolic disorders were included (EFO:0000589). In addition, information on known clinical pathogenicity was downloaded from the ClinVar database14 (downloaded from ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/tab_delimited/variant_summary.txt.gz).
The pipeline software was implemented in bash and R 3.4.4. VCFtools v0.1.1515 was used to filter SNPs from the VCF input file. SNP locations were downloaded from the dbSNP 151 database ((downloaded from ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh37p13/VCF/common_all_20180423.vcf.gz). All coordinates are in GRCh37/hg19 build. SNP coordinates were converted to bed format using awk. dbSNP identifiers were converted to genomic coordinates using dbSNP 151 reference (see above). Bedtools v2.28.016 was used to identify overlap of SNP coordinates with individual annotation sources. Finally, R merge was used to join tables by variant location.
The output file ("final_table.txt") contains SNP coordinates, positionally-overlapping genes, associated clinical significance from ClinVar, associated GWAS trait and p-value, coordinates and state name of overlapping open chromatin states in gut tissues, and name of tissue and genes for significant eQTL associations. The pipeline then filters this file to report only those SNPs with GWAS hits that achieve genome-wide significance (p < 5×10-8); this file is titled "GWASsignficant.txt", and creates a third file with the list of unique genes that meet this criterion ("GWASsignificant_genes.unique.txt"). The user may filter this data still further to identify SNPs with known clinical pathogenicity, as well as SNPs in functionally annotated non-coding regions.
For our test case, we used the SVAI Undiagnosed-1 Patient whole genome sequencing data provided for the Undisclosed-1 hackathon event, hosted by SVAI/Research to the People. We applied our pipeline to chromosomes 1 to 22 and X, Y chromosomes. Out of 6,248,584 SNPs, we identified 151 high-priority variants, overlapping 129 genes, which demonstrate strong evidence for functional significance (Table 3). Therefore, this pipeline allows the user to prioritize certain variants for further downstream analyses, or clinical follow up.
Information includes mapped genes, disease associations from genetic studies, eQTL associations from GTEx, and overlap with open chromatin regions in gut tissues.
This pipeline will allow easy integration of several epigenomic functional annotation maps that could assist in SNV prioritization for clinical and basic research applications. Our provided software can be customized by someone with basic bioinformatics or scripting expertise to generalize to other tissues profiled in the GTEx and NIH Roadmap Epigenomics project.
One beneficial extension would be the identification of variants predicted to affect gene splicing. Such predictions are available in databases of splicing variants, such as dbscSNV17,18, or simple splice site prediction algorithms such as MaxEntScan19,20; the latter uses sequence motifs and maximum entropy calculations for its predictions. This approach is limited by the quality of variant databases, and by models that are limited to predicting only in instances that follow canonical rules for splice site regulation. Another promising avenue to predict aberrant splicing is SpliceAI21, a deep learning-based model that predicts splice junctions from an arbitrary pre-mRNA transcript sequence. Another valuable addition would be that of using higher-order chromatin interaction maps, which allow the mapping of distal regulatory elements, such as enhancers, to genes (e.g. 8).
This article is based on research that occurred at the Undisclosed-1 hackathon event, hosted by SVAI/Research to the People.
The patient provided written informed consent for data release of their medical records, including genetic results, blood work and clinical laboratory reports, to the organisers of the event (SVAI/Research to the People). This consent included data release to participants of Undisclosed-1 during the event, and subsequently for this data to be hosted by SVAI/Research to the People in an online data repository with restricted access (see details in “Data Availability”).
The patient provided written informed consent for the publication of all articles based on the research that was carried out at Undisclosed-1 and any accompanying images.
Since the medical records are the patient’s property, the patient was fully informed of what data release would entail and written informed consent was obtained for the release of the medical records. No ethical approval was sought for the Undisclosed-1 event or publication of articles relating to this event.
Synapse repository: SVAI, Undiagnosed-1 (syn:20554923), https://doi.org/doi:10.7303/syn2055492322.
License information: Since the data contains detailed medical records, access is restricted in order to protect the identity of the patient. Intermediary data is provided throughout the article. In order to access the data, applicants must be registered users of synapse.org and must provide a proposal detailing what the data will be used for. Applicants will also be required to sign a statement that ensures that the data are not shared with others who have not applied to use the data from the SVAI. Please submit applications for data access to [email protected].
Software for this pipeline is available at: https://github.com/shraddhapai/SNPNotes
Archived release at time of publication is located at: http://doi.org/10.5281/zenodo.335227623.
License: MIT
Data in this project contains:
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)