Keywords
dermatomyositis, myositis-specific autoantibodies
dermatomyositis, myositis-specific autoantibodies
Dermatomyositis (DM) is an inflammatory myopathy with characteristic skin manifestations, the pathologies of which are considered autoimmune diseases. DM is a heterogeneous disorder with various phenotypes, including myositis, dermatitis, and interstitial lung disease (ILD)1. Recently, in addition to the already-established anti-aminoacyl-transfer RNA synthetase (ARS) antibody, a number of myositis-specific autoantibodies—including anti-melanoma differentiation-associated protein 5 (MDA5) antibody and anti-transcriptional intermediary factor 1γ (TIF1γ) antibody—that are not detected in patients with an inherited muscle disease2 have been identified. These autoantibodies not only are highly disease-specific but also are associated with distinct clinical features (Table 1)3,4. This article reviews their epidemiology and characteristic clinical features, with a focus on their characteristic cutaneous manifestations, to determine the systemic pathologies of the different types of antibody-associated DM.
| Autoantigen | Clinical features | Typical cutaneous manifestations |
|---|---|---|
| MDA5 | Clinically amyopathic DM* with ILD†, especially rapid progressive ILD | Palmar violaceous macules/papules due to vascular injury |
| TIF1 | Juvenile DM*; cancer-associated DM* | Severe cutaneous manifestations |
| Mi2 | Classic DM* | Sometimes refractory |
| ARS | Anti-synthetase syndrome with chronic ILD† | Mechanic’s hands |
| NXP2 | Juvenile DM and adult DM | Calcinosis |
| SAE | Clinically amyopathic DM* followed by severe myositis including dysphagia | Extensive rash, sometimes as erythroderma |
Anti-MDA5 antibody has a high specificity for clinically amyopathic DM (CADM) presenting rapidly progressive ILD (RP-ILD)5. Anti-MDA5 antibody was first reported as an anti-CADM-140 antibody that reacted with a 140-kDa cytoplasmic protein6 subsequently identified as the retinoic acid-inducible gene I (RIG-I)-like receptor MDA5/IFIH1 (interferon [IFN] induced with helicase C domain protein 1). The anti-MDA5 antibody is detected at high frequencies among patients with DM in Asia (15.8% [26/165 cases] in Japan and 36.6% [53/145 cases] in China)7 and South America (16% [21/131 cases] in Brazil)8 and at low frequency (2.8%, 21/748 cases) in a cohort of patients with DM in a combined European cohort in which 87.4% of enrolled cases were Caucasian9. Case series studies reported CADM frequencies of around 40% in anti-MDA5 antibody-positive patients with DM7,10. A meta-analysis of 16 studies estimated pooled sensitivity and specificity of anti-MDA5 antibody for RP-ILD of 77% (95% confidence interval [CI] 64–87%) and 86% (95% CI 79–90%), respectively, with a pooled diagnostic odds ratio of 20.41 (95% CI 9.02–46.20)11. The severity and prognosis of RP-ILD in anti-MDA5 antibody-positive patients with DM were strongly correlated with anti-MDA5 antibody titer (detected by established enzyme-linked immunosorbent assay) and serum ferritin level12. In a series of 44 Japanese patients with juvenile DM (JDM), 41% were positive for anti-MDA5 antibody13 compared with 7.4% of 285 patients with JDM in the UK14. Both studies reported anti-MDA5 antibody to be strongly associated with ILD; however, only 8 (18%) of the 44 Japanese cases and none of the UK patients developed RP-ILD. A recent cohort study in the UK observed low myositis severity scores depending on muscle biopsies in 11 anti-MDA5 antibody-positive patients15.
The anti-TIF1 antibody was originally described as anti-155/140 and anti-p155 antibodies targeting a 155-kDa nuclear protein, sometimes with a 140-kDa protein16,17. These antigens were subsequently identified as TIF1 family proteins belonging to the tripartite motif (TRIM) superfamily, TIF1γ (TRIM33) and TIF1α (TRIM24), respectively. Anti-TIF1γ antibody was detected in both adult DM and JDM patients and was closely correlated with malignancies, especially in elderly patients18–20, at high risk of dysphagia21 and at low risk of ILD, Raynaud phenomenon, and arthritis/arthralgia22. Anti-TIF1γ antibody was present in 7 to 15% of patients with DM9,23. A meta-analysis including 1,962 patients with DM demonstrated a prevalence of malignancy-associated DM of 0.41 in patients with anti-TIF1γ autoantibody (95% CI 0.36–0.45). The diagnostic odds ratio of cancer was 9.37 (95% CI 5.37–16.34) with low heterogeneity—Cochran’s Q, 14.88 (degrees of freedom = 17, P = 0.604), I2 = 0%—in the presence of anti-TIF1γ autoantibody24. In contrast, 30% of patients with JDM present anti-TIF1γ antibody17,25 and do not develop malignancies.
Patients with anti-ARS antibodies, including anti-Jo-1, anti-PL-7, anti-PL-12, anti-EJ, anti-OJ, anti-KS, anti-Ha, and anti-Zo, share characteristic clinical symptoms such as myositis, ILD, arthritis/arthralgia, Raynaud’s phenomenon, and fever; thus, the term “anti-synthetase syndrome” is also used26.
The anti-Mi-2 antibody is directed mainly to Mi-2β, a component of the nucleosome-remodeling deacetylase complex27. Anti-Mi-2 antibody was detected in 3% of patients with JDM25 and 12% of patients with adult DM9. Anti-Mi-2 antibody-positive patients have a lower risk of ILD and typically respond well to therapy, although the recurrence of DM symptoms is possible23.
The anti-nuclear matrix protein 2 (NXP2) antibody, originally termed anti-MJ antibody, was first identified in a cohort of patients with JDM/juvenile polymyositis (JPM). Generally, anti-NXP2 antibody-positive myopathy is related to either DM or polymyositis (PM) phenotypes. Cohort studies have detected anti-NXP2 antibody in 22 to 25% of patients with JDM25,28. Another cohort study reported that severe myopathy characterized by muscle contractures and atrophy was associated with anti-NXP2 antibody-positive JDM28. In contrast, anti-NXP2 antibody was detected in only 2.3% of patients with adult PM/DM9. Moreover, two cohort studies of patients with adult PM/DM in Japan and the US suggested a possible association between anti-NXP2 antibody and malignancy19,29.
The anti-small ubiquitin-like modifier activating enzyme (anti-SAE) antibody, which was observed in about 6% of patients with DM9, is associated with inflammatory myopathy with extensive rash and dysphagia30,31. The target autoantigen is a heterodimer of SAE1 (40 kDa) and SAE2 (90 kDa). ILD and malignancies were observed in, respectively, 42 and 21% of 46 previously reported patients with anti-SAE antibody-associated DM31.
Myositis-specific autoantibodies are likely to be associated with distinct cutaneous manifestations (Table 1). In the case of anti-MDA5 antibody-associated DM, cutaneous ulceration due to vascular injuries was related to rapidly progressive ILD32,33 and palmar violaceous macules/papules32,34, in which vasculopathy in the medium and small dermal vessels was frequently observed32.
Severe cutaneous manifestations, including V-neck sign, shawl sign, heliotrope rash, Gottron’s papules/sign, and flagellate erythema, are often observed in patients with anti-TIF1γ antibody-associated DM16,17. Fiorentino et al. termed these characteristic cutaneous manifestations palmar hyperkeratotic papules, psoriasis-like lesions, and hypopigmented and “red on white” telangiectatic patches22.
Mechanic’s hands, characterized by keratotic erythema on the sides of the thumbs and forefingers35, are generally specific to patients with anti-synthetase syndrome, including those with anti-ARS antibody-associated DM26.
Juvenile and adult myopathy patients positive for anti-NXP2 antibody have a high risk of calcinosis36, although patients positive for anti-NXP2 antibody include those with JPM/PM. In contrast, anti-SAE antibody-positive patients with DM demonstrated extensive rash, including erythroderma with “angel wings” sign31.
The histopathological findings of cutaneous lesions in DM include vacuolar degeneration of the basilar keratinocytes, lymphocytic inflammatory infiltrate around the dermal blood vessels, and interstitial mucin deposition. We previously analyzed the histological findings of finger lesions characterized according to myositis-specific autoantibodies (anti-ARS, anti-MDA5, and anti-TIF1γ)37. Our study included finger skin specimens from 30, 19, and 25 cases positive for anti-ARS, anti-MDA5, and anti-TIF1γ antibodies classified according to cutaneous histopathological classifications—(i) interface dermatitis, (ii) psoriasiform dermatitis, (iii) eczematous reaction, and (iv) vascular injury—and also analyzed by immunohistochemistry to detect myxovirus resistance A (MxA) expression, which is usually associated with type I IFN activity. Finger eruptions of anti-ARS antibody-positive DM were histologically characterized by not only interface dermatitis but also psoriasiform dermatitis and eczematous reaction, which were rarely observed in the other patients with DM. Dyskeratotic cells were frequently observed in anti-ARS antibody-positive DM, while vascular injury in the upper dermis was found in anti-MDA5 antibody-positive DM. MxA expression in the epidermis was high in anti-MDA5 antibody-positive DM and rarely observed in anti-ARS antibody-positive DM. The conclusion is shown in Figure 1. MxA expression was rarely observed in the muscle biopsy samples. Previous studies also identified anti-synthetase syndrome as a histological subset in muscle biopsy samples among patients with idiopathic inflammatory myositis, which was characterized by perifascicular necrosis38 and negative MxA expression, which is generally highly expressed in the muscle fibers of patients with DM39. Moreover, a recent study reported that plasma IFN-α levels and the expression of IFN-inducible molecules from peripheral blood mononuclear cells and skin biopsies were higher in anti-MDA5 antibody-associated DM patients than those in anti-ARS antibody-associated or autoantibody-negative DM patients40. Collectively, our findings indicate that these histological characteristics are shared between skin, muscle, and blood samples of patients with DM; that anti-ARS antibody-positive patients are clearly distinguished from other DM subgroups; and that the pathogenesis of anti-MDA5 antibody-associated DM is mediated mainly by type I IFN.
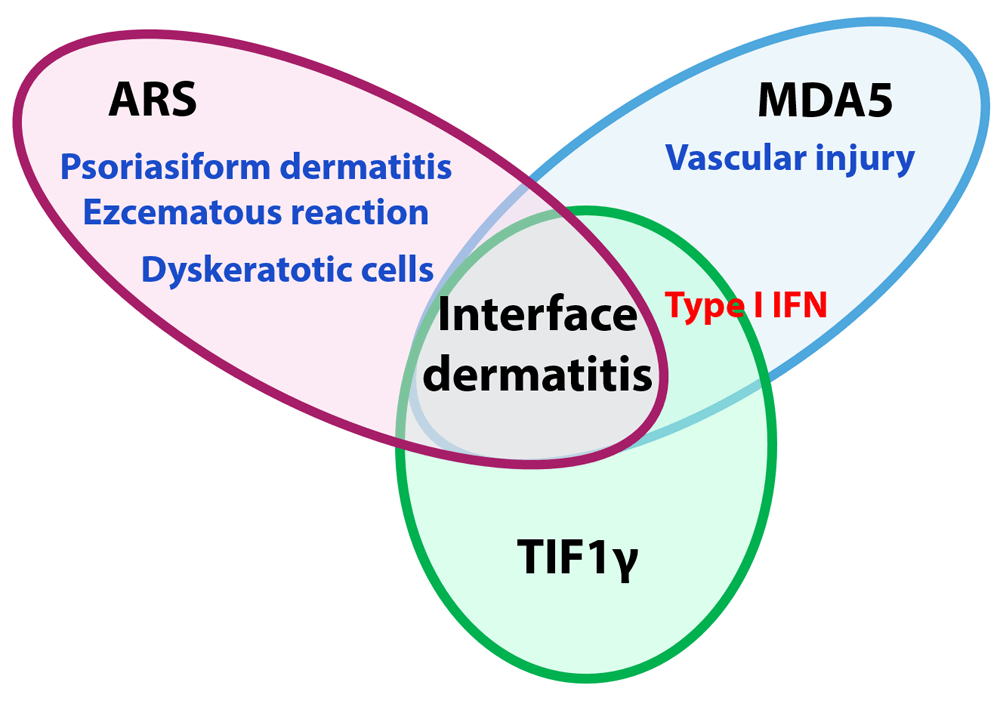
The anti-aminoacyl-transfer RNA synthetase (ARS) antibody-positive dermatomyositis (DM) group is characterized by a mixture of psoriasiform dermatitis and eczematous reaction with interface dermatitis mainly presenting dyskeratotic cells and without epidermal expression of myxovirus resistance A (MxA). Vascular injury in the upper dermis and high epidermal expression of MxA are observed in patients with anti-melanoma differentiation-associated protein 5 (anti-MDA5) antibody-positive DM. Epidermal expression of MxA is also detected in patients with anti-transcriptional intermediary factor 1γ (TIF1γ) antibody-positive DM. IFN, interferon.
Further studies are needed to clarify the differences among the DM subgroups according to myositis-specific autoantibodies and to provide a basis for the development of subgroup-specific DM therapies.
ARS, aminoacyl-transfer RNA synthetase; CADM, clinically amyopathic dermatomyositis; DM, dermatomyositis; IFN, interferon; ILD, interstitial lung disease; JDM, juvenile dermatomyositis; tripartite motif, JPM, juvenile polymyositis; MDA5, melanoma differentiation-associated protein 5; MxA, myxovirus resistance A; PM, polymyositis; RP-ILD, rapidly progressive interstitial lung disease; SAE, small ubiquitin-like modifier activating enzyme; TIF1γ, transcriptional intermediary factor 1γ; TRIM; NXP2, nuclear matrix protein 2
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)