Keywords
MVID, malabsorption, epithelial transport, diarrhea, trafficking
MVID, malabsorption, epithelial transport, diarrhea, trafficking
In 1978, Davidson et al. presented a case report of five infants with persistent severe diarrhea from birth and marked abnormalities of absorption associated with failure to thrive, leading to death in four infants1. The common histological abnormalities in duodenal mucosa from those infants were villus atrophy, crypt hypoplasia (without an increase in mitoses or inflammatory cell infiltrate in the lamina propria) and absence of a brush border in villus enterocytes, and an increase in lysosome-like inclusions2,3. Originally referred to as Davidson's disease, congenital microvillus atrophy, and intestinal microvillus dystrophy, the disease was named microvillus inclusion disease (MVID) in 1989 by Cutz et al.4.
As with all rare genetic diseases, the diagnosis of MVID was quite challenging until recently and required histological evaluation for confirmation. It is important to note that MVID has a very low incidence, making it extremely difficult to investigate its pathophysiology5. The morphological anomalies observed in the enterocytes of patients with MVID are widely utilized in disease diagnosis2. Until recently, the gold standard in diagnosing MVID was combined light and electron microscopy of small bowel biopsy samples of patients. The abnormalities are mainly observed in the small intestine and less frequently in the colon6. However, some studies have shown that the colon and rectum biopsies may also contain characteristic features which would be useful in diagnosing MVID2,6,7.
The key hallmarks which aid in the differential diagnosis include blunted or absent microvilli, accumulation of secretory granules, and microvillus inclusions (MIs) in the epithelial cells2,8. As depicted in Figure 1, these granules, in most cases, are positive for periodic acid Schiff (PAS) stain and CD10 with an intracellular PAS or CD10 positive line in enterocytes that is commonly detected. Another apical marker which may aid in the identification of the trademark MIs is villin, an apical surface marker of enterocytes9. An important factor which should be accounted for during histological evaluation of biopsies is sampling variability and patient-to-patient variability. The diagnosis is confirmed further by genetic testing, which can specifically identify the genetic anomaly of each patient. In this instance, currently, there is a registry which tracks each genetic variation observed in MVID patients to facilitate ease of access to patient-related data for clinicians involved in the management of this rare genetic disorder10.
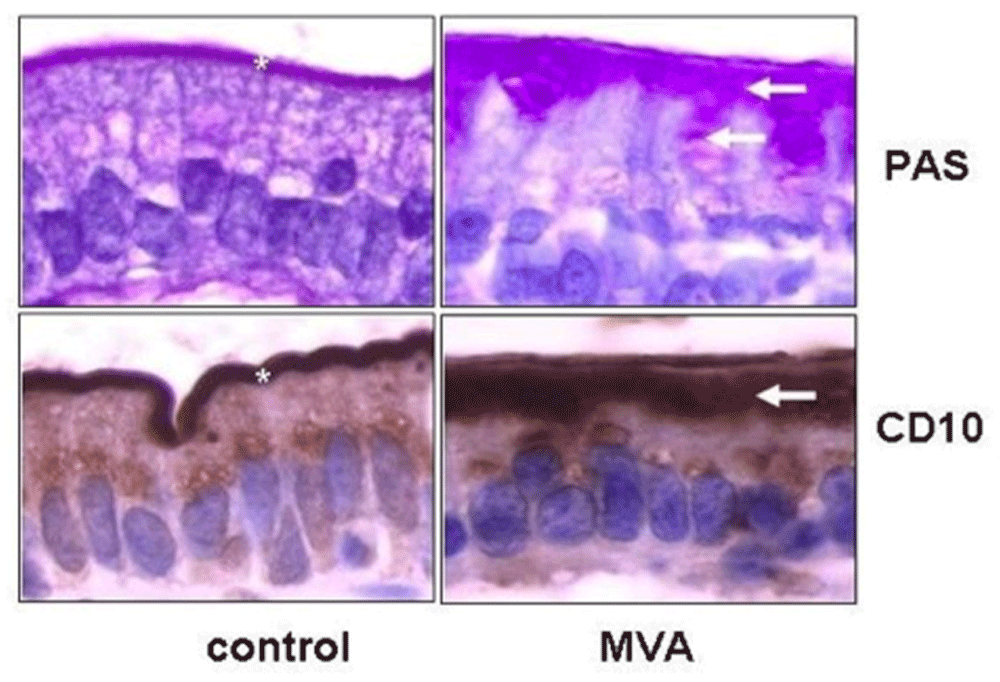
MVA, microvillous atrophy; PAS, periodic acid Schiff
There are several features that differentiate MVID from other diarrheal conditions with similar clinical presentation including the onset at birth, absence of inflammation, presence of vacuoles containing granules with the characteristic PAS and CD10 positive stain observed under light microscopy, and presence of MIs (Table 1). Other congenital disorders such as chloride and sodium diarrhea can be easily excluded from biochemical assays or genetic testing6,11. Tufting enteropathy is a disorder with similar onset and blunted villi; however, the presence of surface apical tufts as opposed to apical inclusion bodies in enterocytes distinguishes tufting enteropathy from MVID. Enteroendocrine cell dysgenesis can be differentiated from MVID by the lack of enteroendocrine cells and the presence of normal microvilli. Finally, abetalipoproteinemia is distinguished from MVID by the presence of fat vacuoles and a foamy cytoplasm20.
| Congenital disease | Major gene/s mutated | Distinctive feature(s) |
|---|---|---|
| Microvillus inclusion disease | MYO5B12, STX313, STXBP214 | Blunted microvilli, microvillus inclusions |
| Chloride diarrhea | SLC26A3 or Down Regulated in Adenoma (DRA)15 | High-chloride diarrhea (fecal Cl– >90 mM/L) and normal microvilli |
| Sodium diarrhea | SPLINT2 (serine peptidase inhibitor 2), GUCY2C (guanylate cyclase C), and SLC9A3 (sodium hydrogen exchanger 3 (NHE3)16 | High-sodium diarrhea (fecal Na+ >145 mM/L) and normal microvilli |
| Tufting enteropathy | EPCAM (epithelial cell adhesion molecule)17 | Presence of surface apical tufts with blunted villi |
| Enteroendocrine cell dysgenesis | NEUROG3 (neurogenin-3)18 | Lack of enteroendocrine cells with normal villi |
| Abetalipoproteinemia | MTTP (microsomal triglyceride transfer protein)19 | Fat vacuoles with foamy cytoplasm and normal villi |
Earlier studies in patients with MVID showed high stool volume (150 to 300 mL/kg/day) with remarkably elevated sodium content (approximately 100 mmol/L)2,21. The diarrhea present in MVID is considered to be non-osmotic in nature (i.e. fecal ion gap <100 mOsm) and is persistent even when the patient is unfed22. This type of diarrhea is categorized as electrolyte transport-related diarrhea caused by mechanisms involving net secretion of anions (chloride, bicarbonate, or potassium) and/or net inhibition of sodium or chloride absorption11,23–25. Steatorrhea and impaired glucose absorption have also been reported in MVID patients26,27. Various studies have shown mislocalization of apical membrane-targeted proteins such as sucrase isomaltase, alkaline phosphatase, and sodium hydrogen exchanger 3 (NHE3) in MVID, which might partly explain the pathophysiology of malabsorption and diarrhea28. Due to the high-volume and persistent diarrhea observed in these patients, the main life-saving treatment option remains life-long total parenteral nutrition (TPN). The use of life-long TPN poses many complications including sepsis and worsening cholestatic liver disease that may require intestinal transplantation. However, outcomes of intestinal transplantation remain poor2,27. Owing to the immature nature of the enterocytes present in infants with MVID, absorption of essential nutrients is hampered and, therefore, recent studies are directed at developing therapeutic agents which are capable of increasing the maturity of the enterocytes to ultimately recuperate the loss of absorptive capacity of the small intestine29. Although little progress has been made in developing treatment options, the growing research has certainly highlighted relevant mechanisms linking perturbation in cellular trafficking and signaling pathways to functional physiological defects leading to malabsorption and chronic diarrhea30.
The identification of gene mutations linked to trafficking pathways in MVID has paved the way for further research into better understanding of this intricate and challenging enteropathy. The major mutation observed in MVID patients is in the MYO5B gene, the key molecular motor gene regulating trafficking of important proteins into the brush border of the intestinal epithelial cells31. An online registry for MVID patients and their mutations has been generated which currently has 188 MVID patients10. Although the majority of MVID patients exhibit mutations in MYO5B, mutations in other genes have also been identified that present with less severe enteropathy. For example, mutations in soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein syntaxin-3 (STX3) cause a variant form of MVID with lateral microvilli and occasional microvillus occlusions13. In addition, patients with mutations in STXBP2, encoding the syntaxin-binding protein-2 (MUNC18-2) protein, also have intestine-related hallmarks of MVID besides their primary diagnosis of familial hemophagocytic lymphohistiocytosis type 5 (FHL5), a hyper-inflammatory immune disorder14. Recent studies by Dhekne et al.32 provided further evidence that MYO5B, STX3, and STXBP2 genes are functionally linked in MVID patients. In this regard, analysis of subcellular distribution of STX3 and MUNC18-2 in enterocytes of intestinal biopsies from patients with MYO5B or STXBP2 mutations showed that MUNC18-2 and STX3 accumulated in intracellular puncta in the enterocytes of MVID patients as compared to apical localization in brush border plasma membrane in control enterocytes. In addition to the native biopsy samples, in vitro Caco2 model epithelium has been used extensively to recapitulate the loss of MYO5B on epithelial polarity and intracellular trafficking. Interestingly, MYO5B knockdown mimicked the loss of apical microvilli and lack of polarity and was associated with internalization of several apical membrane transporters such as Na+/H+ exchanger NHE331,33,34 and Down Regulated in Adenoma (DRA)34. While both NHE3 and DRA localization were significantly reduced on the apical membrane of human MVID enterocytes and MYO5B knockdown (MYO5B-KD) C2BBe cells, the localization of cystic fibrosis transmembrane conductance regulator (CFTR) was mostly preserved28. Functional studies confirmed that Forskolin-stimulated CFTR ion transport was intact in MYO5B-KD T84 cells28.
Another recent study using stable MYO5B-KD in CaCo2-BBE cells established the critical role of MYO5B interactions with specific RAB small GTPases (RAB8A and RAB11) in MVID35. MYO5B-KD cells showed loss of microvilli; however, no MIs were observed. The expression of WT MYO5B in MYO5B-KD cells restored microvilli, while the expression of MYO5B–P660L, an MVID-associated mutation found within the Navajo population (that cannot bind to RAB11A), induced the formation of MIs but did not rescue the MYO5B-KD phenotype. On the contrary, the expression of a RAB8A binding-deficient MYO5B mutant partly restored the microvilli loss, but no inclusions were formed. These studies demonstrated that the disruption of the MYO5B–RAB11A interaction results in the formation of MIs, whereas MYO5B–RAB8A binding is important for microvilli formation35. Recent studies by Vogel et al. identified Rab11- and/or Rab8-positive recycling endomembrane compartments that were enriched with apical membrane proteins, including STX3 and NHE3, in MVID patients’ enterocytes36.
With respect to mechanisms underlying the origin of inclusions and microvillus loss, a recent review by Schneeberger et al.29 highlighted three potential models or a combination of these models to explain the pathological hallmarks of MVID. In the first, described as a trafficking model, defects in vesicle trafficking caused by MYO5B or STX3 mutations result in the subapical accumulation of vesicles and in the lack of appropriately polarized apical proteins. In the second model (recycling model), perturbations in the recycling and delivery of apical recycling endosomes (AREs) result in the subapical accumulation of apical proteins and in the formation of microvilli-containing macropinosomes. As discussed above, MYO5B is required for the localization of RAB11A-positive AREs, which contain various signaling molecules, such as pyruvate dehydrogenase kinase (PDK1), protein kinase C (PKCi), and serine threonine protein kinase (MST4) colocalized with ezrin28,32,37. The third local induction model proposes that in MVID, RAB11A-positive AREs accumulate and function as a subapical signaling platform to induce ectopic intracellular microvillus formation37. The presence of MIs in MVID is the pathognomonic finding based on microscopy of intestinal tissues in diagnosing patients. However, the formation of these inclusions in enterocytes is not yet defined as a cause or consequence of the disease, although the latter is more accepted in the current clinical setting. Plausibly, MIs may represent a secondary effect of overall disrupted epithelial polarity in MVID38.
In the first report of animal models of MVID initiated about 4 years ago, in 2015, Schneeberger et al. and Cartón-Garcia et al. described the deletion of the MYO5B gene in mice and its close phenotypic similarity to the human disease39,40. The inducible intestine-specific knockdown of MYO5B could successfully recapitulate human MVID in just 4 days post induction. However, germline knockdown of MYO5B in mice very closely showcases hallmarks of MVID in the duodenum during the gestational stage (day 20 of gestation) and in newborn mice40. In addition, in a recently developed swine model published as an abstract form, where the mutated gene in MYO5B (P663L) is introduced, the disease phenotype is similarly discernable41. The pig model is the first large animal model of human MVID that develops diarrhea shortly after birth and may be useful for preclinical studies.
Similar to studies in cell lines and patients with MVID, intestinal tissues from MYO5B-knockout mice showed decreased localization of apical protein NHE3 but not CFTR42. Also, the tamoxifen-inducible VilCreERT2;MYO5Bflox/flox model demonstrated a loss of apical NHE3, sodium glucose transporter-1 (SGLT1), DRA, and aquaporin-7 (AQP7)38. These mice did not show an intestinal barrier defect, based on Ussing chamber analysis, but exhibited decreased SGLT1 activity and increased CFTR activity. However, in MVID patient intestinal explants, increased permeability has been reported43. Also, mislocalization of CFTR was demonstrated in some patient biopsies34. These differences further highlight that knockout of myo5B may not necessarily resemble the presence of a mutated MYO5B protein. In addition, it is unclear if these models have defects in the large intestine, as most of the studies have utilized the small intestine alone.
Intestinal enteroids have recently emerged as an important model which closely recapitulates the human disease phenotype due to epithelial defects. Because of the presence of all types of epithelial cells and the self-renewing capacity of the enteroids, these cultured native intestinal epithelial cells represent a superior model as compared to cancer cell lines. In this regard, there is a significant scarcity of patient-derived enteroids from MVID13. This is mainly due to the lack of a reasonably large patient cohort and the very early onset and fatality of the disease. However, intestinal enteroids generated from different mouse models where MYO5B is knocked down exhibited abnormalities with features similar to those seen in the small intestinal tissues of MVID patients33,38,42. A recent study conducted by Mosa et al. underscored the importance of studying the pathology of MVID by demonstrating the ability to rescue the defects present in MUNC18-2 (mutated in FHL5) knockdown mouse enteroids by expressing the human WT protein and not by the mutant FHL5 patient variant (P477L)36,44. It is noteworthy to mention that owing to the rare nature of the enteropathy, long-term preservation of patient samples to generate organoids is warranted to enhance the current understanding of the disease.
Although very simple, consisting of only a few enterocytes, the C. elegans nematode model possesses a close resemblance to human intestinal epithelium with distinct polarization of apical and basolateral membranes with a prominent microvillus brush border. In this regard, by silencing various components in the V-ATPase complex (an important regulator of cellular trafficking), the authors identified that specific subunits of the protein complex, in particular V0, are upstream of other genetic defects which leads to a MVID-like phenotype in this model45. Due to the simplicity of the model, this may be important for use as a platform to study the development of the disease as well as potential cellular mechanisms, which can be a target for developing drug molecules for MVID management.
The MYO5B gene is expressed in all epithelial tissues, but the most prominent phenotype is observed in the intestine. However, several extraintestinal pathologies have also been reported in other tissues. In this regard, pathologies identified include renal Fanconi syndrome, cholestasis, hematuria, and pneumonia27,46. Therefore, animal models of MVID could be useful to study these conditions that may be missed in humans owing to the complications associated with disease diagnosis, the very early onset, and lack of survival. With respect to biliary dysfunction, a recent study found cholestasis in 30% of their patient cohort, which was characterized by a low level of serum gamma-glutamyl transpeptidase (GGT)47. The study reported abnormalities in the recycling of MYO5B and RAB11A and mistargeting of bile salt export pump (BSEP) to the canalicular membrane of hepatocytes. Although cholestasis in MVID patients was previously thought to be solely due to TPN-related toxicity, evidence has emerged supporting cholestasis in the absence of TPN due to apical trafficking defects in MVID hepatocytes48. In this regard, the investigators noted that the unexpected low levels of GGT in MVID patients contrasted with the high levels of this surrogate in cases of liver failure associated with TPN. In a very recent preliminary study conducted in MYO5B null mice and pigs with Navajo mutation (published in an abstract form), the authors demonstrated an interference with apical membrane trafficking in hepatocytes. Specifically, multidrug resistance associated protein-2 (MRP2) and BSEP were mislocalized to subapical compartments. In addition, dipeptidyl peptidase-4 (DPPIV) enzyme was mistrafficked and the liver bile canaliculi lacked branching, highlighting the importance of MYO5B in studying liver dysfunction associated with MVID patients49.
Malabsorptive disorders lead to retarded growth and nutritional deficiencies. The complex nature of these disorders poses a challenge for treatment options50. Understanding the pathophysiological mechanisms of malabsorption should improve current management protocols and immensely enhance our knowledge regarding intestinal physiology. In this regard, increased understanding of the intriguing malabsorptive disorders of childhood such as MVID should offer new insights at the cellular and molecular levels to unravel the link between cellular trafficking and epithelial absorptive processes. The research in the field of MVID has considerably progressed over the last decade. The generation of novel mouse models with MYO5B deletion has been successful in recapitulating various hallmark features of MVID. So far, the utilization of these models has not only substantiated the role of MYO5B and trafficking machinery in the disease’s pathogenesis but also underscored the importance of cellular trafficking mechanisms in maintaining optimal function of nutrient and electrolyte transporters such as SGLT1 and NHE3. Unlike the in vitro and in vivo mouse models, where loss of MYO5B ideally disrupts intracellular trafficking in all cells, the manifestation of abnormalities in MVID patients is patchy and sometimes confined to a few enterocytes5. In addition, although some studies described the presence of abnormalities in the colon and rectum of MVID patients, most animal models focused only on the duodenum and upper small intestine35,38,39,42. More studies in the distal parts of the small intestine and colon should broaden our understanding of the compensatory mechanisms that the intestine may employ to adapt in consequences of MYO5B mutations. The mechanisms underlying lipid malabsorption associated with MVID remain elusive. Therefore, investigations to explore the molecular basis for dysregulation of lipid absorption in MVID patients and mouse models are warranted. The inducible MYO5B-deficient mouse models have the additional advantage of studying the consequences of time- and age-dependent occurrences of disease-specific hallmarks29,33,38. Although MVID is a rare disorder, the organoids derived from MVID patients can provide unique opportunities to model the disease and modify the mutated genes by state-of-the-art approaches, including the CRISPR/Cas9 gene editing system, for rescuing the defective phenotype29.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)