Keywords
Active TraR site, Quorum Sensing, Molecular Docking, 3D-QSAR
This article is included in the Cheminformatics gateway.
Active TraR site, Quorum Sensing, Molecular Docking, 3D-QSAR
The intracellular communication mechanism known as quorum sensing (QS) makes it easier for bacteria to develop cooperative behaviors, which coordinate their activities in order to function as a multicellular unit1. QS is a process based on the synthesis, diffusion, and perception of small signal molecules called autoinducers (AIs), through which a bacterial population can monitor their cell density and regulate specific gene expression in response to local changes in its population density2,3. Bacterial behaviors regulated by QS include the secretion of virulence factors, biofilm formation, bioluminescence, sporulation and conjugation4.
QS in Gram-negative bacteria is mediated by N-acyl homoserine lactones (AHLs). In this AHL-dependent system, the QS signal is detected by a cytosolic transcription factor (R). AHL synthase (Protein I) produces AHL molecules that can diffuse through the cell membrane and as they reach a critical extracellular concentration, they enter the cell again and bind to the R protein, forming the R-AHL complex, which activates the expression of some specific genes5,6.
The Gram-negative bacterial species Agrobacterium tumefaciens regulates the expression of the genes necessary for the replication and conjugation of the tumor-inducing plasmid in dicotyledonous Ti plants through the QS system, causing the disease known as ‘crown gall’ that occurs in greenhouse and fruit crops7. Its QS system is homologous to LuxI /LuxR by Vibrio fischeri and is made up of the autoinducer synthase, the TraI protein, the N-(3-oxo-octanoyl)-L-homoserine lactone (3OC8HSL) autoinducer and the transcriptional receptor, the TraR protein5. This communication system is activated when the lactones synthesized by TraI at high population densities accumulate until reaching a certain level and when they reach that concentration threshold, they are detected by TraR. The TraR-3OC8HSL complexes activate the transcription of the target genes in the Ti plasmid, including those required for conjugation8. In effect, the TraR protein is a transcriptional regulator in the QS mechanism of A. tumefaciens.
Since QS signaling is vital for the regulation of bacterial gene expression that has an impact on numerous microbial physiological functions, including those related to pathogenicity in humans, plants and animals, there is enormous interest in the design and implementation of QS system inactivation strategies9. One of the strategies for interruption of the QS is the blockade of the transduction of the QS signal, which can be achieved by the presence of an AHL antagonist capable of competing or interfering with the binding of the signal molecule to its LuxR receptor10.
For this purpose, we combined two molecular modeling strategies: molecular docking and three-dimensional quantitative structure-activity relationship (3D-QSAR). First, the molecular docking methodology was applied to a series of 34 analogues of AHL on the TraR transcriptional receptor to simulate the binding of analogues at the active TraR site. Secondly, analysis of the structural characteristics and interactions of the ligands was carried out in order to evaluate how the inhibitory activity is affected by its properties. Finally, 3D-QSAR models were generated to describe the correlation between the experimental biological activity using partial least squares (PLS) calculations and steric and electrostatic properties, which theoretically predict the activity of the 34 analogues of AHL through statistical parameters and evaluate the prediction of the models obtained.
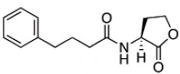
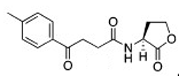
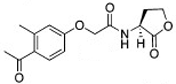
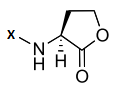
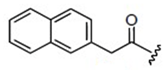
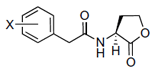
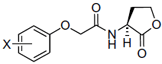
The chemical structures of 34 AHL analogues were taken together with their half maximal inhibitory concentration (IC50) biological activity from Geske et al. (Table 1)11,12. These structures were optimized using the theory of functional density with the functional X3LYP, together with the DGDZVP calculation base13,14, using the Gaussian09 program15 (as an open-source alternative, GAMESS could be used16).
AHL, acyl homoserine lactone; PHL, phenylacetyl homoserine lactone; POHL, phenoxyacetyl homoserine lactone; PPHL, phenylpropionyl homoserine lactone. Structures and IC50 values for each analogue were obtained from Geske et al. 200711.
Molecular docking calculations were performed to predict the binding modes among the 34 analogues of AHL and the TraR receptor. The 3D structure of the TraR receptor and its native co-crystallized ligand 3OC8HSL were downloaded from the Protein Data Bank, with the identifier 1L3L. The complex in PDB format was visualized and prepared using the structure preparation tool available in the Sybyl X 2.1.1 package17 and the Amber force field was used to assign the partial atomic charges of the protein. As an alternative, the open-source program, Avogadro could be used18. The native ligand and all water molecules were removed from the complex, hydrogen atoms were added and side chain amides and imidazoles were fixed (protonated), assuming a physiological pH of 7. The structure of extracted native ligand was optimized with the same conditions as the 34 analogues, mentioned above.
Once the structure of the native ligand and the TraR receptor was obtained, the docking protocol was initiated in the molecular simulation program AutoDock 4.219. The preparation of both the protein and ligand was performed using the graphical user interface of AutoDock, called AutoDockTools20. This preparation involved the addition of hydrogen and the assignment of Gasteiger charges21. After this, the potential maps for each type of atom in the ligand were calculated, executing the grid parameter using the Autogrid program incorporated in AutoDock. In this study, the grid box was delimited, centered on the TraR binding site, with dimensions 50x50x50 Å and spacing between points of 0.375 Å. Finally, for the conformational search, the Lamarckian genetic algorithm was chosen and for the validation 500 conformations were registered, with the remaining parameters used as per the default setting. With these parameters, docking validation was carried out by re-coupling the native ligand to the binding site of the TraR receptor.
Once the docking protocol was validated, and its ability to reproduce the interactions that promote protein-ligand binding confirmed for this particular case, TraR-3OC8HSL, the docking calculations for the 34 AHL analogues and the receptor in question were continued following the previous protocol, with the only difference being that 200 conformations were recorded for each analogue, of which the best were chosen according to their binding energy values and their position within the binding site of the TraR protein. The interactions of the selected protein-ligand complexes were analyzed on the Protein-Ligand Interaction Profiler webserver22.
Two 3D-QSAR models were constructed from structural alignments. The first model was constructed by taking the structure of the ligand, with the best inhibitory activity, reported by Geske et al. 200711, as a template. The other model was constructed based on the 34 best-selected poses of each analogue-TraR molecular docking23, which generated an automatic alignment24. The comparative molecular field analysis (CoMFA)25 descriptor was obtained by using the QSAR tool implemented in Sybyl X 2.1.1. Steric and electrostatic field energies were calculated by using the Lennard-Jones and Coulomb potentials, respectively, with a dielectric constant 1/r based on the distance of all interactions. To calculate the steric and electrostatic energies, we used the force field of Sybyl X.2.1.1, Tripos26. As an alternative, the open-source program, Open3DQSAR could be used27.
For the evaluation of the 3D-QSAR models obtained, the cross-validation technique was used with the ‘leave-one-out’ method. With this validation, the predictive power of the models was determined and, in this way, the best model was determined. An exploratory analysis of the correlation between molecular fields and biological activity was also performed using PLS regression; thus, the correlation coefficients (R2) of each model were obtained using the SAMPLS algorithm of Sybyl X 2.1.1. Finally, the cross-validation was done, giving the values of Q2 and R2CV. According to these values both models were compared, and it was deduced which had the best predictability.
In order to guarantee the reliability of the protocol used for molecular docking among analogues of AHL and the TraR receptor, validation of the search algorithm and scoring function combination was performed by re-docking the native ligand to the binding site of the TraR receptor using the molecular docking approach. From the large number of conformations generated, the conformations that best reproduced the position and interactions of the native ligand within the binding site were chosen. This conformation was compared with the co-crystallized structure of the same native ligand, as is depicted in Figure 1. In the alignment of the conformations of the native ligand in (green color) and the resulting docking (pink color), it can be seen that the coupled structure located at the TraR binding site adopts a conformation almost identical to the co-crystallized structure of the native ligand and in this way, both the lactone ring and the acyl side chain is located suitably at the receptor binding site.
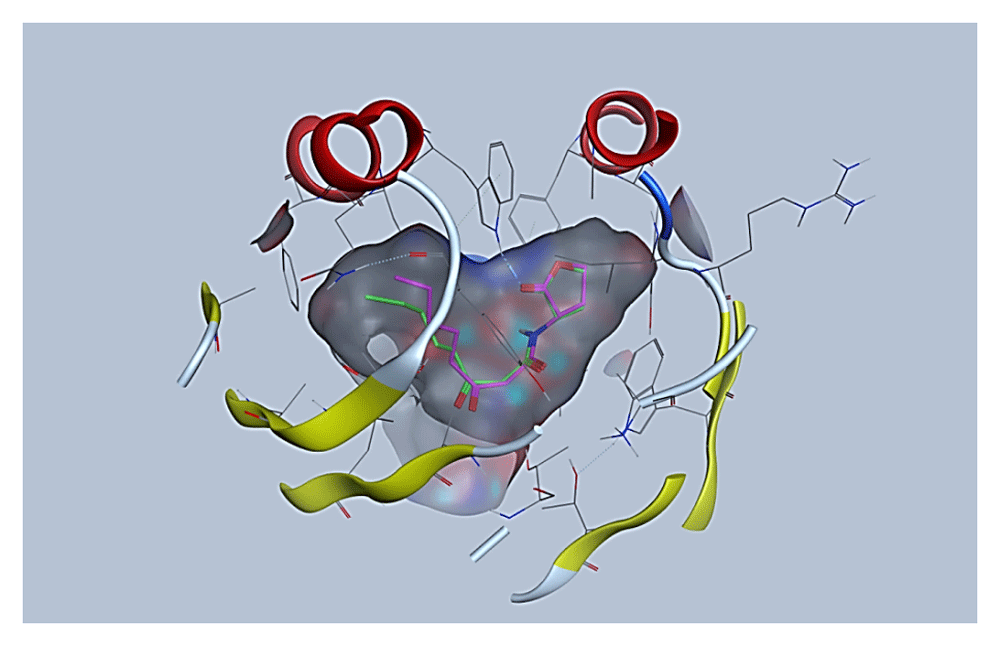
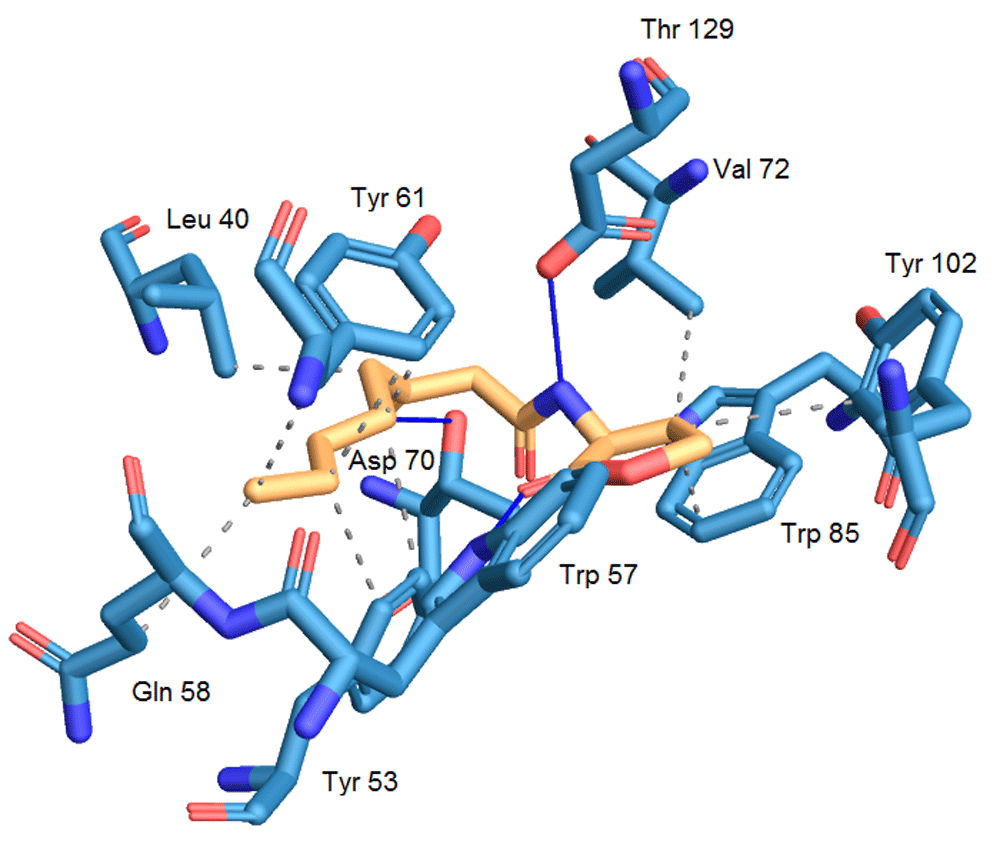
The mean quadratic root deviation (RMSD) value calculated by AutoDock was used to confirm whether the chosen conformation was correct or not. The RMSD value of the chosen conformation is 0.56 Å; generally, an RMSD value of less than 1.5 or 2 Å is considered to be optimal28. In Figure 2, the interactions established in the conformation resulting from the molecular docking are visualized. In this ligand-receptor complex, the majority of hydrogen bond interactions that contribute to the binding of the native ligand and TraR are reproduced, as well as the correct folding of the TraR receptor29. These interactions include hydrogen bonding between the Trp 57 residue and the carbonyl group of the lactone ring, hydrogen bonding between Asp 70 and the amino group and a bond between the second carbonyl group and Thr 129, with the exception of the bond between the Tyr 53 residue and the first carbonyl group located in the side acyl chain. Hydrophobic residues also contribute to the stable binding of the native ligand with TraR by hydrophobic interactions with residues Leu 40, Tyr 53, Tyr 61, Val 72, Trp 85. The interactions with Thr 51, Phe 62 and Ile 102 do not occur in said conformation.
The structural interactions of this group of molecules were classified into five groups according to the differences present in the acyl chain reported by Geske et al.11,12. This classification was made taking into account the importance of the influence of the variation of the acyl chain on the interactions that form the 34 analogues of the AHL within the TraR binding site. In addition to the classification by groups, the individual structure of each analogue was divided into two regions, A and B, as is shown in Figure 3, the first consisting of the lactone ring and the carbonyl group and the second region comprising the acyl chain, which is the one that varies between the different analogues. In this way, the analysis of the interactions is facilitated since each analog tends to form mostly the same interactions in the region A, unlike those formed in region B, which tend to be different30.
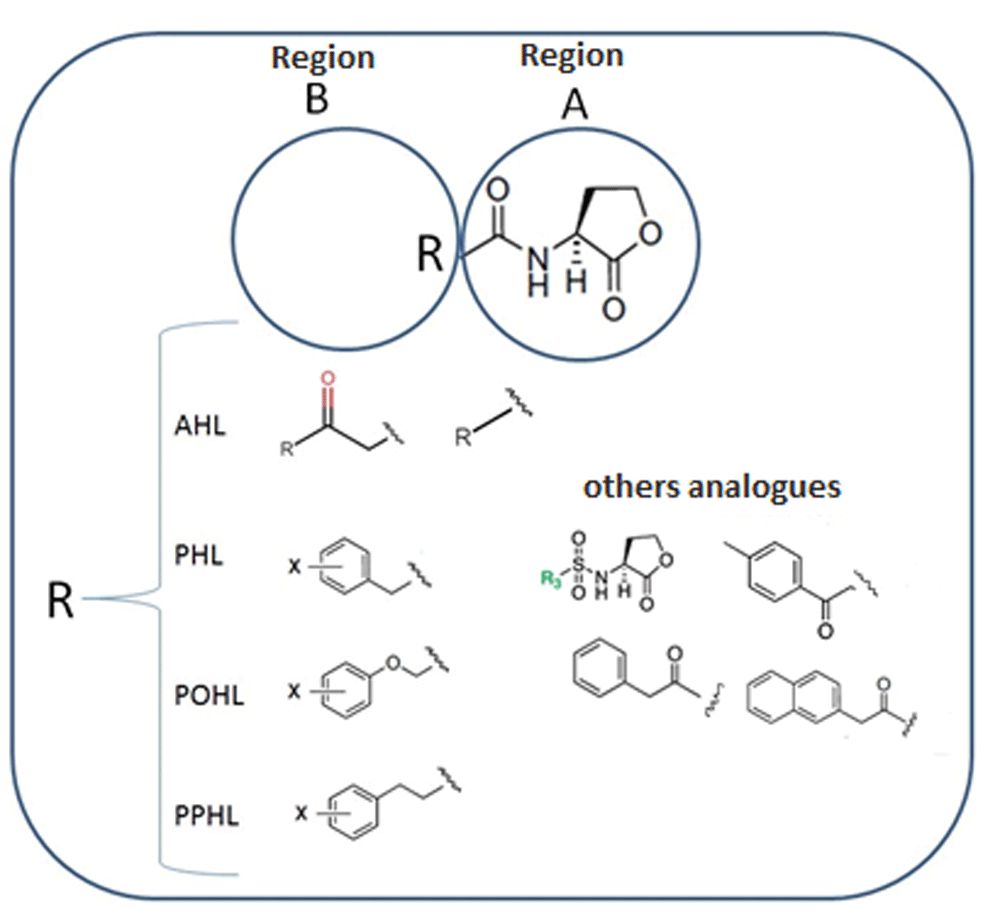
Region A is formed by the lactone ring and the amide function and region B has variable side chains.
For each analogue-TraR docking, two or more clusters were generated. In these clusters, the different ways of binding can be identified. From the analysis of the clusters of each analogue-TraR docking, the best 34 conformations were chosen using the orientations that reported the lowest binding energy as the criterion of choice. The analogue-TraR complexes chosen, in comparison to 3OC8HSL, are, in general, similarly oriented at the TraR receptor binding site. The structure of these analogues was constructed based on the structure of homoserine lactone, with variations in the acyl chain of the native ligand. Table 2 shows the IC50 values and the free energies calculated using AutoDock for all 34 ligands.
Ligands 4, D6, C10, E22 and E35 have the highest values of inhibitory activity (IC50), so the analysis of their structure-activity relationship for each group was taken as precedents. Analogues 4, C10, E22 and E35 form two hydrogen bonds with the TraR receptor; one between the carbonyl group of the lactone ring and the amino group in the Trp 57 residue and the second between the carbonyl group of the first carbon of the acyl chain and the residue Thr 129. Ligand D6 also forms these two hydrogen bonds, but also forms another with the Asp 70 residue and the carbonyl group of the acyl chain. The native ligand forms the same three hydrogen bonds mentioned that contribute to the bond at the TraR protein binding site. Similarly, the remaining ligands form these bonds but with the absence of one or two of them and differences in bond distances. The interactions of ligands D6 and E35 are detailed in Figure 4.
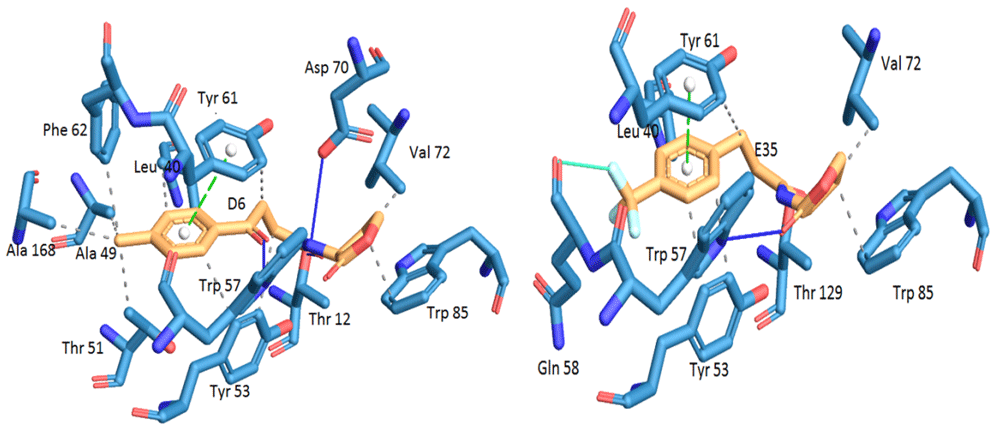
Hydrogen bonds are shown in blue, hydrophobic interactions in gray, π-π stacking in green and halogen bonds in cyan.
On the other hand, the possible hydrophobic interactions formed by these ligands in the hydrophobic pocket of TraR involve both the lactone ring and the acyl chain. Structurally, ligand 4 is classified within the group of non-native AHLs with an acyl chain of seven carbon atoms; one less than the native ligand 3OC8HSL. This side chain allows the formation of eight possible hydrophobic interactions with Ala38, Leu40, Gln58, Phe62 and two interactions with each residue of Tyr61 and 63. In addition, in the lactone ring are the interactions with Val72 and Trip85, for a total of ten interactions. The D6 ligand has in its structure a side chain a carbonyl group on carbon 4 and a toluyl group. This structural modification places it within the group of other analogues, and between this chain and the protein it is possible to form 11 hydrophobic interactions; eight with the same residues as ligand 4 and also those formed with Ala49, Thr51 and Ala168. The C10 ligand has in its side chain a phenylacetanoyl group and E22 a phenoxyacetanoyl group, which places them in the phenylacetyl homoserine lactones and phenoxyacetyl homoserine lactones groups, respectively. Both form six hydrophobic interactions; four in the side chain with Leu40 and Tyr53 with different distances of connection, and two in the lactone ring with Val 72 and Trp85. Finally, of the 34 analogues, ligand E35 reported the highest inhibitory activity against TraR. It is classified in the phenylpropionyl homoserine lactones group since it has the phenylpropionyl group in its side chain and its possible hydrophobic interactions are the same as those mentioned for C10 and E22.
The differences among the possible hydrophobic interactions mentioned above for the different ligands representing each group and the interactions of the native ligand in the hydrophobic pocket of TraR are notable. However, these are not the only differences; some analogue-TraR complexes reported other possible interactions, such as π-π stacking, which are favored in ligands that in their structure possess an aromatic group, as can be seen in the complexes of ligands D6, C10, E22 and E35. The formation of this interaction occurs between the benzene rings of these molecules and the Tyr 61 residue. This type of interaction is also reported in other ligands, but with the amino acid Tyr53.
On the other hand, analogues that have halogen atoms in their side chains can form other types of interactions called halogen bonds. In this study, ligands C10, D15, E21, E23, B7, and E35 have halogen bond interactions with the Gln 58 residue and the E23 ligand with Trp 57. Studies of the characteristics of this type of halogen bond establish that they are strongly directional and the distance between the halogen and the nearby electronegative atom should ideally be less than the sum of its Van der Waals radii. Moreover, although the fluorine atom is a halogen, it does not have the capacity to form these interactions due to its great electronegativity31. According to the above, the halogen bonds of ligands D15, E23 and E35 are discarded as they are not possible since the interaction occurs between a fluorine atom and an oxygen atom. The sum of the van der Waals radii for the remaining ligands C10, E21 and B7 are calculated as follows: in the C10 ligand where its iodine atom and the Gln 58 oxygen interact, this value is 3.50 Å; in the E21 ligand where its bromine atom and the Gln 58 oxygen interact, the sum of the radii is 3.37 Å; and in ligand B7 where its bromine atom and oxygen of Gln 58 interact, the sum of the radii is 3.37 Å. The respective distances between the atoms bound for each ligand-receptor complex are 3.90 Å , 2.14 Å and 2.06 Å. Comparing these values, it follows that the only halogen bonds that are shorter than the value of the sum of the radii of the bound atoms are those of ligands E21 and B7 with the amino acid Gln 58.
It is quite clear that the addition of certain groups such as phenyl and halogenated substituents can generate other interactions, which could confer changes in the increase or decrease in the inhibitory activity of these analogues against the TraR receptor. These extra interactions (halogen bond, π-π stacking), formed by the analogues and the TraR receptor, are not present when TraR is in complex with the native ligand.
For ligand-based alignment, ligand E35 was used as a template since it was the ligand with the highest inhibitory activity against TraR and for the receptor-based alignment, the best poses generated in the molecular docking, that is, those that reported the lowest binding energy and the correct orientation in the active site of the receptor, were used. Figure 5A and 5B show the superposition of all ligands for the ligand-based model (Model 2) and receptor-based model (Model 1), respectively.

A) Model 1 and B) Model 2.
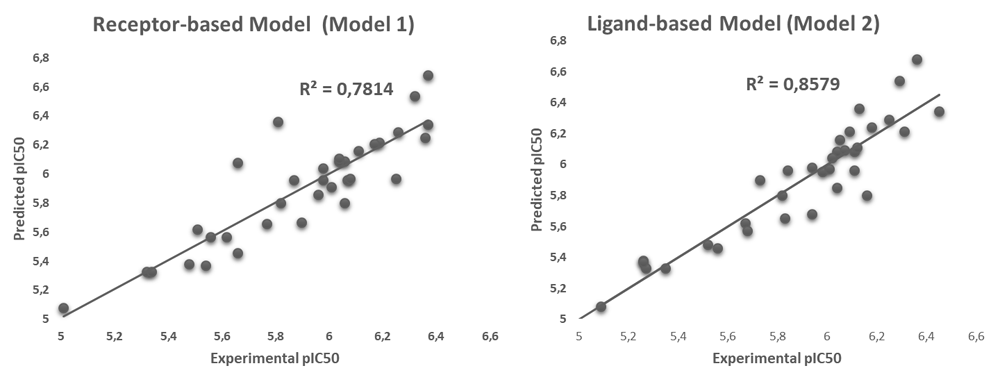
The correlation analysis between CoMFA values and in vitro biological activities of the 34 analogues was performed by PLS and the evaluation of the predictive capacity of the models generated through the leave-one-out cross-validation method. Q2 values of 0.457 and R2 are obtained with cross validation of 0.856 for the Model 2, and a Q2 of 0.697 and R2 with cross validation of 0.781 for the Model 1. In effect, Model 1 shows a lower correlation so that their statistical confidence is lower than in model 2. Table 3 shows the other results of both PLS and cross-validation analyzes.
Figure 6 shows correlation of models 1 and 2, it can be seen that in model 2 there is a better pattern of adjustment of the points along the trend line, which translates into the prediction of biological activity values closer to those reported experimentally, unlike the graph for Model 2 where the residual dispersion is greater. From these comparisons, the Model 2 is statistically profiled as the one with the best correlation and the greatest predictive power. Even so, the Model 1 should not be discarded altogether since it also takes into account the structural similarity between the analogues and the native ligand; the position and orientation taken by the ligands at the receptor binding site according to the conformational freedom allowed by the amino acid residues that comprise it.
Through the interpretation of the contour maps obtained by CoMFA, it is possible to know which structural characteristics of the ligands correlate with the changes in their biological activity values. These contours are represented by colors; steric contour maps are represented in green (region favorable for bulky groups) and yellow (region not favorable for bulky groups), while those of red (region favorable for electronegative substituents) and blue (region favorable for electropositive substituents) correspond to electrostatic contours32.
In Figure 7 the contour maps of the CoMFA models calculated are shown, taking as an illustrative example those generated for ligand D15, which records one of the highest values of inhibitory activity against TraR in the phenoxyacetyl homoserine lactones group. In these contours, it can be observed that the sterically favored regions are in positions close to the fluorine atoms found in the benzene ring substituent in the ligand structure in both Figure 7A and 7B. This is because this region has high electronegativity, which favors interactions with residues that have partially positive charges in their structures, while the sterically unfavorable regions, in yellow, spread around the phenyl group. However, on most of the hydrogen atoms in the phenyl ring, the positively charged regions that favor the electrostatic field are identified, as can be seen in more detail in Figure 7B. In the same way, carbonyl and ether groups with a negative charge are seen as electrostatically favorable regions due to the negative potential of oxygen atoms. In addition, the existence of halogens promotes the formation of halogen bonds with residues containing oxygen atoms in their structure. All these groups mentioned are identified as regions that would contribute to the antagonistic activity of ligand D15 against TraR.
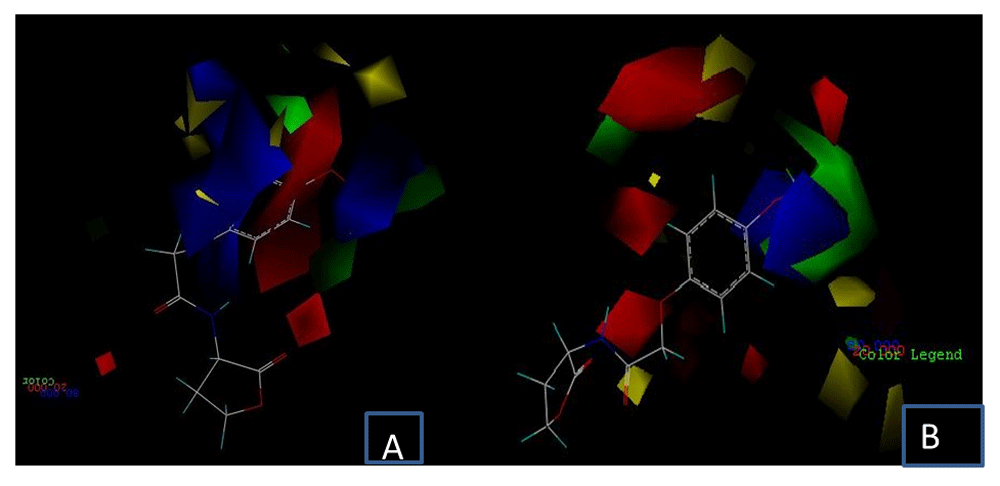
A) receptor-based model and B) ligand-based model. Steric field: green: sterically favorable; yellow: sterically unfavorable. Electrostatic field: red: negative potential favorable; blue: negative potential unfavorable.
In most of the 34 ligand-receptor complexes obtained by molecular docking, the formation of hydrogen bonds and similar hydrophobic interactions with the same amino acid residues were observed. The most common hydrogen bonds are established with Trp 57, Thr 129 and Asp 70. The most recurrent hydrophobic interactions are formed with Ala 38, Leu 40, Tyr 53, Tyr 61, Val 72, Trp 85. Certain structural modifications such as the presence of aromatic rings and halogen atoms allow the formation of other interactions such as molecular type π-π stacking and halogen bonds. From the above, it is possible to affirm that there is a marked relationship between the type of interactions and the activity of the ligands, where the differences with those formed by the native ligand may improve the inhibitory activity of the analogues against TraR.
The PLS statistical analysis of each model and subsequent internal validation allowed comparison of the predictive capacity under parameters such as R2 and Q2, analyzing how the predicted inhibitory activity values vary when the experimental biological activities are correlated with the properties of the optimized structures of the ligands (model based on the ligand) and, for the model based on the receptor, when the same experimental activities are correlated with the properties of the poses that the ligands adopt in the molecular docking. This resulted in the best model in statistical terms being the model based on the ligand. Even so, the receptor-based model, since it is based on the simulations of the analogue receptor binding, provides information based on the position and orientation that the analogues can adopt according to the chemical nature of the TraR receptor's active site, unlike the model based on the ligand, which is based solely on optimized structures.
Regarding the study of the relationship of the structural characteristics that influence the values of biological activity, the 3D-QSAR-CoMFA models suggest that it is possible to improve the inhibitory activity of new ligands by adding groups with positive and negative charges of favorable electrostatic potentials such as carbonyl groups, ether groups, halogens, and in some cases the nitro substituents and the phenyl group, which favors the formation of hydrophobic interactions. Bulky groups attached to the phenyl ring and other groups such as sulfur are identified as unfavorable regions for the design of new analogues with greater inhibitory activity against TraR.
Figshare: Receptor_Alim_2019_Pose_E_D_1.mdb (1).zip. https://doi.org/10.6084/m9.figshare.10255739.v123.
This project contains the following underlying data:
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.


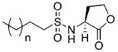
Comments on this article Comments (0)