Keywords
Sarcoidosis, treatment, endpoints, corticosteroids, fibrosis, quality of life
Sarcoidosis, treatment, endpoints, corticosteroids, fibrosis, quality of life
Sarcoidosis is a multisystem granulomatous disease of unknown cause. Granulomas consist of conglomerations of inflammatory cells, predominantly lymphocytes and macrophages. In sarcoidosis, these granulomas may deposit in any organ but most commonly develop within the lung, where they are found in more than 90% of sarcoidosis cases1. The decision to treat pulmonary sarcoidosis is complex because (a) the disease may cause neither symptoms nor permanent organ damage2; (b) a small percentage of cases, maybe 10 to 20%, are progressive or cause fibrosis (that may result in permanent lung dysfunction, significant morbidity, and even mortality) or both3; (c) there are no reliable biomarkers to predict whether the natural course of pulmonary sarcoidosis will be benign or lead to severe impairment4; and (d) treatment of pulmonary sarcoidosis with corticosteroids and other therapies is associated with significant toxicity5. These issues force clinicians, clinical researchers, and pharma to carefully select appropriate pulmonary sarcoidosis patients who have the potential to receive impactful benefits from interventions and to select the appropriate methods to assess therapeutic responses. Clinical trial design for pulmonary sarcoidosis has been problematic, and major trials have been criticized as not adequately accounting for the natural history of the disease in order to determine the benefits of study drugs6. This article will explore the indications for treatment of pulmonary sarcoidosis and methods to assess the effects of therapy. This exercise will involve the construction of a proposed conceptual framework outlining how pulmonary sarcoidosis leads to outcomes that are clinically impactful to patients or clinicians or both. This conceptual framework will suggest that patients in pulmonary sarcoidosis trials require partitioning into disease profiles that each require distinct clinical endpoints to optimally assess the effects of therapy.
Because sarcoidosis may affect any organ in the body and its severity is highly variable, it is problematic to define specific criteria for treatment. Most sarcoidosis clinicians agree with Athol Wells’s distillation of the treatment indications for sarcoidosis to just two: situations of danger and significant quality of life (QOL) impairment (“Wells’s law”)7.
Sarcoidosis is a systemic disease, and situations of danger are not confined to the lung. Cardiac sarcoidosis may cause sudden death, eye involvement may result in blindness, and neurosarcoidosis may result in coma and permanent neurologic impairment. In the case of pulmonary sarcoidosis, situations of danger develop in a minority of patients and are listed in Table 1. Dangerous situations from pulmonary sarcoidosis are confined almost exclusively to those who develop fibrocystic disease, which constitutes 10 to 20% of these patients8,9. Interestingly, the treatment of most of these dangerous situations involves therapy other than anti-granulomatous therapy, although most clinicians and sarcoidosis drug trials focus on obliterating the sarcoid granuloma.
The immunopathogenesis of fibrosis in sarcoidosis is not well understood. It is not known whether the fibrosis is triggered by profibrotic inflammatory events, by an inherent predisposition toward fibrosis in a subset of patients, or by an exaggerated wound-healing response to uncontrolled, chronic inflammation10. Mechanisms proposed to be involved in the development of sarcoidosis-associated fibrosis include alveolar macrophage-induced fibrosis11, transition from a T helper 1 to T helper 2 signature12, transforming growth factor-beta13, upregulation of profibrotic genes that may “transmit” signals for fibrosis through the blood compartment14, and M2 polarization of alveolar macrophages15.
It is conjectured that the fibrosis in pulmonary sarcoidosis results from the granulomatous inflammation. This conjecture is supported histologically where the majority of the fibrosis from sarcoidosis develops within or around the granuloma, resulting in a “hyalinized granuloma”16. This conjecture is further supported anatomically through chest imaging studies17 (Figure 1) as well as analyses of explanted end-stage fibrotic lungs of pulmonary sarcoidosis patients undergoing lung transplantation demonstrating that fibrosis is distributed in a lymphangitic pattern, similar to the usual location of pulmonary granulomas16,18. Finally, this conjecture is also supported by biomarkers of granulomatous inflammation in that a report of 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography demonstrated significant pulmonary FDG uptake in 22 (85%) of 26 patients with fibrotic pulmonary sarcoidosis19. These data suggest that the granulomatous inflammation of sarcoidosis and the development of fibrosis often go hand in hand20. The amount of fibrosis that develops is variable, from negligent to copious amounts. As previously mentioned, a significant fibrotic response from the granulomatous inflammation of sarcoidosis occurs in, at most, only 20% of cases8,9. Herein lies a key dilemma in preventing the granulomatous-induced fibrosis of sarcoidosis: since there is no available biomarker to predict which 10 to 20% of patients with pulmonary sarcoidosis will develop significant fibrosis, does the clinician (a) treat all pulmonary sarcoidosis with anti-granulomatous therapy that will prevent the development of fibrosis in 10 to 20% but subject 80% to 90% to potential drug toxicities from corticosteroids and other medications or (b) withhold anti-granulomatous therapy that will result in serious complications in the 10 to 20% who develop fibrosis? Clearly, a reliable biomarker that can accurately predict the development of significant pulmonary fibrosis from sarcoidosis is an unmet need. Effective anti-fibrotic therapy would also be very useful for this form of sarcoidosis, although none is available at present.
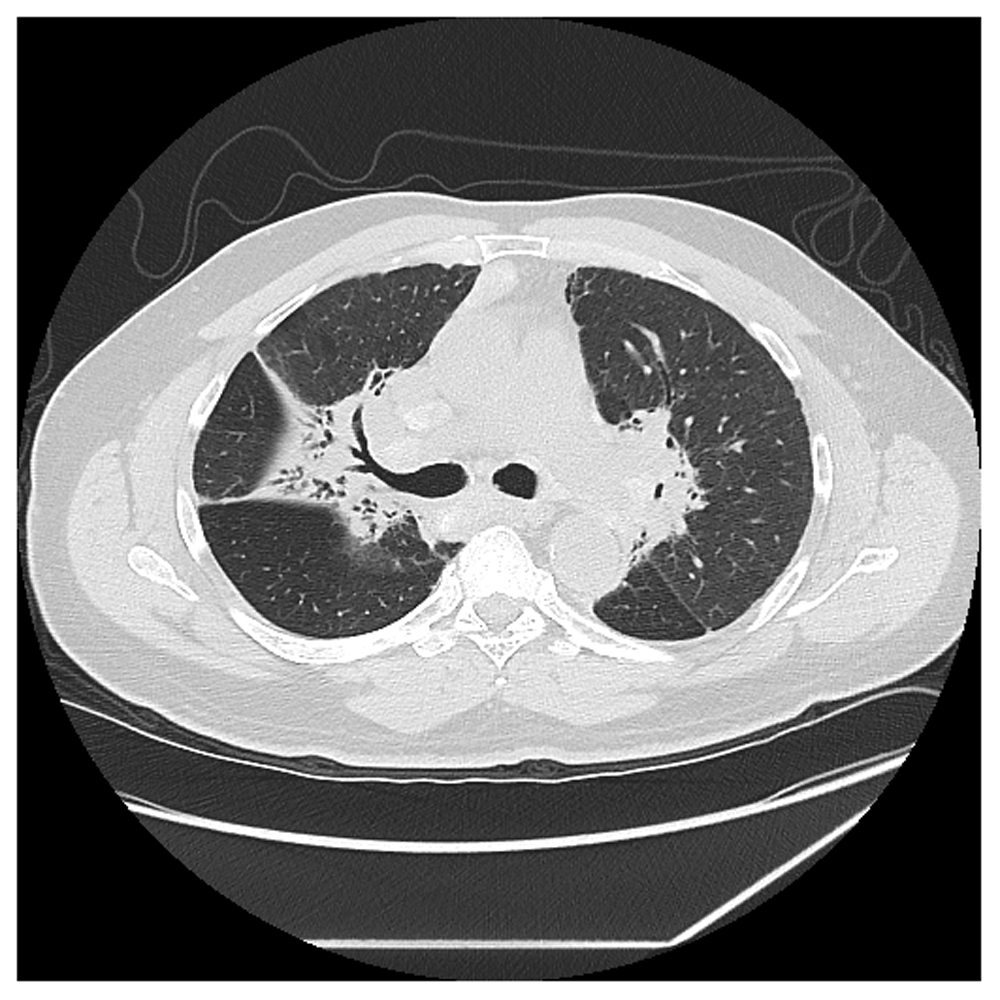
Note how the fibrosis is concentrated centrally along the bronchovascular bundles. This is the same location where the granulomas from pulmonary sarcoidosis tend to deposit. This location of fibrosis provides anatomic evidence that the fibrosis in sarcoidosis is a by-product of the granulomatous inflammation of the disease. We confirm that we have obtained permission to use this image from the patient included in this presentation.
Because the dangerous situations associated with pulmonary sarcoidosis are not common, the more common of the two indications suggested by Wells’s law to treat pulmonary sarcoidosis is sarcoidosis-induced impairment of QOL. A survey of 1842 European patients with sarcoidosis found that QOL and functionality were judged as the most important disease outcomes21. One could argue that sarcoidosis therapy is used primarily to improve physiology or organ function impaired by granulomatous inflammation or to decrease the granuloma burden , but a critical examination of this issue suggests that this is not the case22. Sarcoidosis-induced granulomatous inflammation may not cause a significant physiologic derangement or result in a significant impairment of QOL23. A common clinical situation where this is the case is with asymptomatic bilateral hilar lymphadenopathy24. Even when the granulomatous inflammation of pulmonary sarcoidosis results in physiologic abnormalities, they are often minor8,23,25 and do not invariably lead to appreciable symptoms23,25. In addition, the correlation between pulmonary dysfunction and pulmonary symptoms is poor in pulmonary sarcoidosis26,27. Therefore, the presence of granulomatous inflammation or mild physiologic derangements from pulmonary sarcoidosis does not mandate anti-sarcoidosis therapy unless the patient’s QOL is significantly impaired or significant organ dysfunction occurs22.
QOL assessment in pulmonary sarcoidosis is also significantly impacted by the potential toxicity of therapy, particularly corticosteroids. Corticosteroids are the drug of choice for the treatment of pulmonary sarcoidosis28,29. In most instances, they work quickly and reliably. However, they are associated with a number of potential side effects that may impair QOL. These include weight gain, mood swings/psychological disturbances, diabetes, osteoporosis, hypertension, glaucoma, cataracts, edema, acne, increased skin and vascular fragility, and increased risk of infection. Corticosteroid therapy has been clearly linked to the development of significant toxicities in patients with sarcoidosis30. If the clinician focuses primarily on reducing the granuloma burden or improving physiology, a medication-induced impairment of QOL may be undetected such that the clinician assesses the patient as improved while the patient feels worse off. This scenario may be relatively common, as it has been shown that those receiving corticosteroids at three major university sarcoidosis clinics in the US had statistically and clinically significant reductions in health-related QOL (HRQOL) compared with sarcoidosis patients not receiving corticosteroids27, and those receiving more than 500 mg or prednisone equivalent per year had a statistically and clinically worse HRQOL than those on a lower total yearly dose31.
The assessment of QOL in pulmonary sarcoidosis is problematic. HRQOL refers to the extent that the physical, social, mental/emotional, cognitive, or spiritual domains (or a combination of these) are affected by a medical condition or its treatment32. The HRQOL status of a patient may be assessed informally through unstructured historical questioning. Such an assessment may be thorough and used to make treatment decisions, but in the reality of the current era of patient care where time is compressed, such a detailed evaluation rarely takes place22. For this reason, many have advocated that the assessment of HRQOL in patients with sarcoidosis be assessed by patient-reported outcome measures (PROs), which are patient-administered reports of how they function or feel about their health condition and the effects of therapy33,34. HRQOL PROs provide a quantitative assessment of QOL and functional status in order to assess potential therapeutic effects. In addition, HRQOL PROs may be performed outside of patient clinic visit time and even serially between visits to assess the effects of therapy. However, there are several limitations of using HRQOL PROs on which to base pulmonary sarcoidosis treatment decisions. First, although some sarcoidosis disease–specific PROs have been developed and validated35–37, most HRQOL PROs used in patients with sarcoidosis are derived from other disease cohorts. However, an HRQOL PRO derived from non-sarcoidosis cohorts may be used in sarcoidosis provided that there is no plausible rationale that patients with sarcoidosis would assess the trait being measured in a different way than the cohort in which the PRO was validated23. Second, although PROs can reliably assess HRQOL in large clinical cohorts, most PROs have inadequate resolution to be useful in individual patients. PROs developed with modern methodology, such as item response theory (IRT), may have adequate resolution to accurately assess HRQOL in individuals38–40. Finally, QOL and HRQOL are not synonymous, as many high-priority QOL issues do not pertain to a medical condition or its treatment. It is often problematic to distinguish HRQOL issues from non-health-related QOL issues using HRQOL PROs. Because of these concerns, the role of HRQOL PROs in determining treatment decisions for individual patients with pulmonary sarcoidosis has not been standardized. There is uniform agreement that the clinician should not make treatment decisions in isolation without some insight into the patient’s HRQOL status, preferences, goals, and wishes.
Sarcoidosis may also cause symptoms or organ dysfunction that is not related to tissue deposition of granulomas or the development of fibrosis. These entities are collectively known as parasarcoidosis syndromes41, and they may be responsible for significant QOL and functional impairment. Examples of parasarcoidosis syndromes are erythema nodosum42, small fiber neuropathy43, fatigue syndromes44, pain syndromes45, cognitive decline46, and vitamin D dysregulation47. It has been postulated that these syndromes result from systemic release of mediators associated with the granulomatous inflammation of sarcoidosis43,48. Although these parasarcoidosis syndromes may not result in appreciable pulmonary symptoms, they may be the result of the granulomatous inflammation from pulmonary sarcoidosis. Parasarcoidosis syndromes may respond to anti-granulomatous therapy49,50, although the decision to use such therapy must weigh the benefits versus the side effects of treatment in relation to the impact of the parasarcoidosis syndrome22. However, the etiology of parasarcoidosis syndromes is probably more complex than simply a result of granulomatous inflammation, as there may be several non-granulomatous contributions to some of these disorders, such as pain, fatigue, and cognitive impairment syndromes related to the psychological impact of the presence of a chronic disease or decreased function/mobility or both51,52.
We have proposed a conceptual framework of how pulmonary sarcoidosis impacts patients through the relationships of granulomatous inflammation, physiologic impairment, fibrosis, parasarcoidosis syndromes and the impact of anti-sarcoidosis therapy (Figure 2). This conceptual framework will serve as the blueprint for the selection of the appropriate patients with pulmonary sarcoidosis for therapy and the assessment of such therapy that will be outlined below. Based on this conceptual framework, the treatment of pulmonary sarcoidosis can be directed against three different aspects of the disease: anti-granulomatous therapy for acute untreated disease, anti-granulomatous therapy for chronically treated disease, and therapy for fibrotic sarcoidosis.
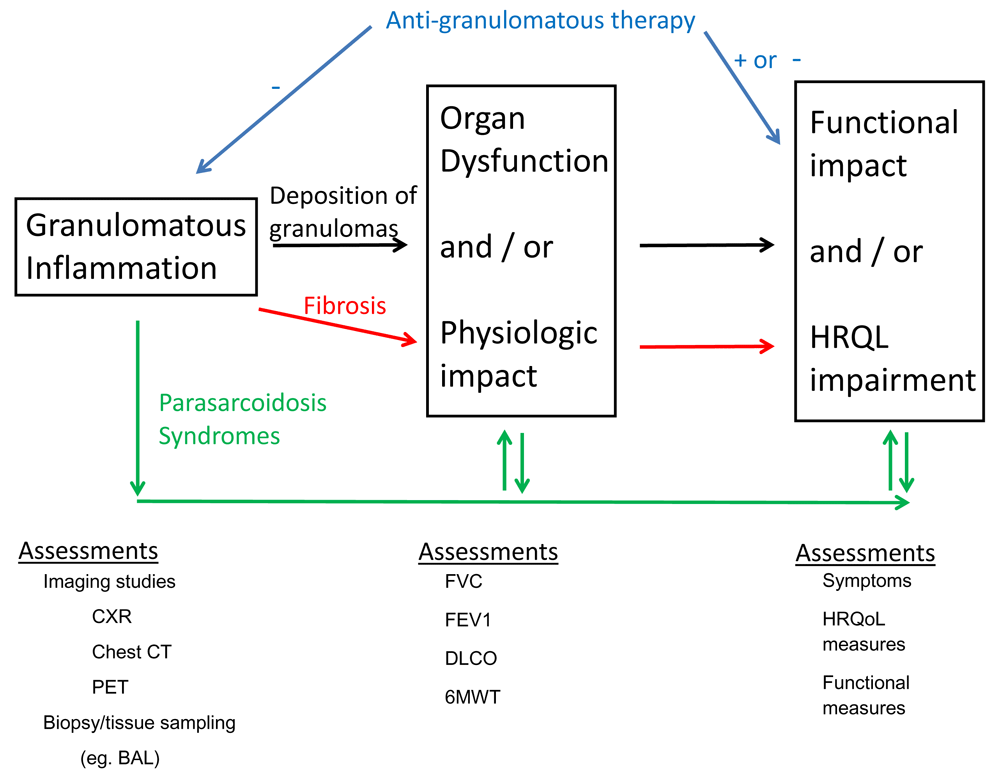
The framework outlines the relationships of granulomatous inflammation, physiologic impairment, fibrosis, parasarcoidosis syndromes, and the impact of anti-sarcoidosis therapy. Assessments refer to methods to assess the processes described within the boxes. Black arrows show the potential clinical progression of pulmonary sarcoidosis directly related to pulmonary deposition of sarcoidosis granulomas. This process is potentially reversible. Red arrows show the potential clinical progression of pulmonary sarcoidosis from pulmonary fibrosis. This process is irreversible. Green arrows show the potential clinical progression of pulmonary sarcoidosis from the development of parasarcoidosis syndromes. These processes may be reversible. Blue arrows show the effects of anti-granulomatous therapy that may reduce granulomatous inflammation (−) and thereby improve health-related quality of life (+). However, the toxicity of anti-granulomatous therapy, particularly corticosteroids, may worsen health-related quality of life (−). 6MWT, 6-minute walk test; BAL, bronchoalveolar lavage; CT, computed tomography; CXR, chest x-ray; DLCO, diffusing capacity of carbon monoxide; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; HRQL, health-related quality of life; HRQoL, health-related quality of life; PET, positron emission tomography.
This conceptual framework is directed at immunologic and physiologic factors of pulmonary sarcoidosis that impact patient health. It reflects relevant issues concerning the evaluation of these drugs in clinical pulmonary sarcoidosis trials, including the establishment of study entry criteria and clinically relevant endpoints. However, issues concerning the management of patients with pulmonary sarcoidosis extend beyond these immunologic and physiologic factors. For example, psychosocial and socioeconomic concerns are important considerations of therapy. Such concerns include the cost of care, including medication costs, access to care, social support, employment issues, emotional and psychological issues, and ease in maintaining a healthy lifestyle.
Acute pulmonary sarcoidosis is a form of the disease that causes worsening pulmonary symptoms and dysfunction over weeks to months and is thought to result from the formation of new granulomas within the lung or growth of existing pulmonary granulomas53. Obviously, anti-granulomatous therapy is a rational approach to the treatment of this condition. As depicted by the black arrows in Figure 2, the sarcoidosis granulomas deposit in involved organs such as the lung and impair normal physiology. These physiologic derangements may lead to functional and QOL impairment. As previously mentioned, the correlations between sarcoidosis-induced granulomatous inflammation and physiologic derangements and between physiologic derangements and QOL impairment are weak.
Obviously, patients with pulmonary sarcoidosis should be considered for anti-granulomatous therapy only if they have evidence of active granulomatous inflammation. Table 2 lists clinical criteria that have been used to determine that granulomatous inflammation is present. These criteria have been used by clinicians to decide when to treat pulmonary sarcoidosis with anti-granulomatous therapy and as entry criteria in pulmonary sarcoidosis trials of anti-granulomatous drugs54–58. However, as previously mentioned, because the granulomatous inflammation of sarcoidosis may not progress or even spontaneously resolve without therapy and therapy is associated with significant toxicity, treatment of acute pulmonary sarcoidosis also requires that the condition has resulted in a significant impairment of QOL.
Figure 3 shows the effect of corticosteroid therapy on pulmonary function for acute untreated pulmonary sarcoidosis. Corticosteroids are most commonly used as initial treatment for acute untreated pulmonary sarcoidosis because they work almost universally and work more rapidly than other medications. If no fibrosis has developed, effective granulomatous therapy should extinguish all granulomatous inflammation and return lung tissue to its normal state, resulting in complete resolution of physiologic impairment. Although resolution of pulmonary physiologic impairment might resolve QOL and functional impairment, this may not be the case because of the potential for the patient to develop numerous corticosteroid side effects. Figure 4 shows a hypothetical relationship between the efficacy of corticosteroids in resolving the granulomatous inflammation of sarcoidosis and the risks of corticosteroid toxicity. Although high doses of corticosteroids are effective in suppressing granulomatous inflammation, they accomplish this by placing the patient at high risk of corticosteroid side effects. The clinician often attempts to locate a “sweet spot” daily dose of corticosteroids where granulomatous inflammation is adequately suppressed without inducing significant corticosteroid toxicity. Because many of the side effects of corticosteroids (for example, weight gain, osteoporosis, and cataract formation) are cumulative, the duration of corticosteroid therapy may affect the tolerable maximum daily dose of corticosteroids that can be safely used. For this reason, it has been advocated that the daily corticosteroid dose be tapered for pulmonary sarcoidosis cases that have responded to therapy. Although intermittent short courses of corticosteroids may be effective in these instances, acute relapses of pulmonary sarcoidosis are very common such that this treatment approach is rarely satisfactory59.
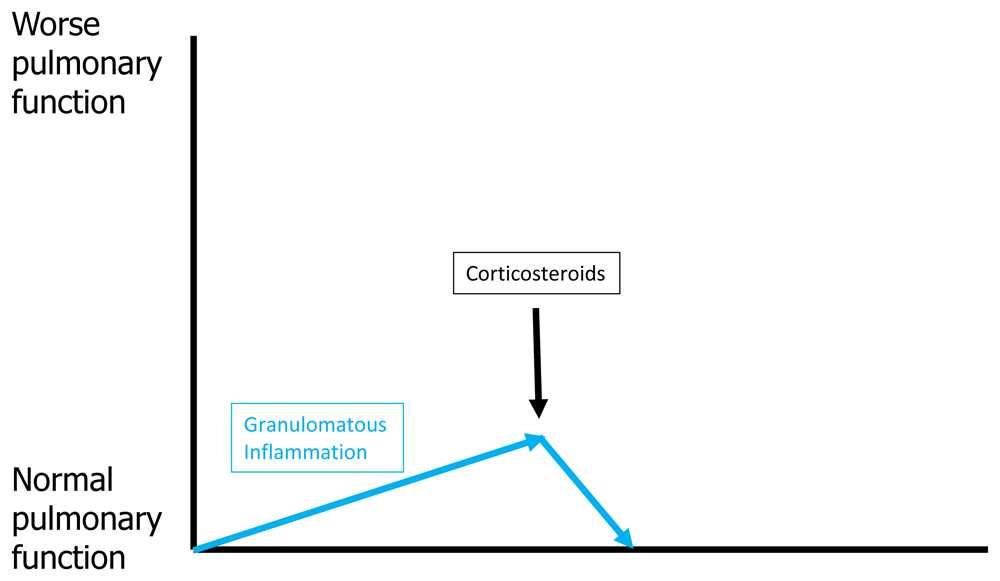
Treatment of the granulomatous inflammation of pulmonary sarcoidosis (blue arrows) with corticosteroids (black arrow) should result in clearance of the deposited granulomas within the lung and return pulmonary function back to normal if no appreciable lung fibrosis has yet developed. It is important to note that, because of potential corticosteroid drug toxicities, such therapy may not resolve quality-of-life issues.
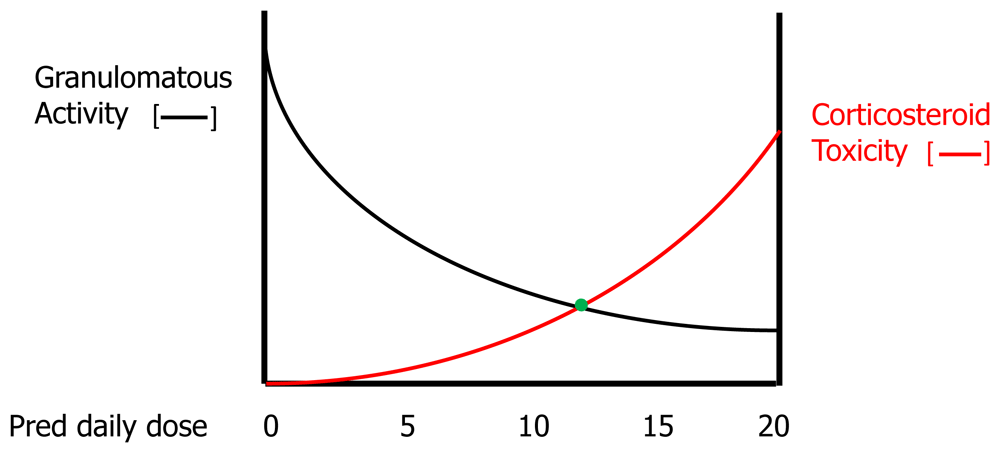
Increasing corticosteroid doses are more effective in resolving granulomatous inflammation at the cost of a greater risk of corticosteroid toxicity. The clinician attempts to locate a “sweet spot” (green dot) where anti-granulomatous efficacy is significant without appreciably raising the risk of corticosteroid side effects.
The endpoints for the treatment of acute untreated pulmonary sarcoidosis should include all three of the following: (a) improvement/resolution of granulomatous inflammation, (b) improvement in pulmonary physiology, and (c) improvement in function status or QOL or both. The rationale for this is explained in Table 3, where it is demonstrated that when any one of these endpoints is not reached, the benefit of treatment of acute untreated pulmonary sarcoidosis is questionable. This does not imply that each clinical pulmonary sarcoidosis trial must incorporate all of these endpoints. For example, if a drug therapy has been previously demonstrated to reliably reduce or eliminate the granulomatous inflammation of sarcoidosis, it may not be necessary to demonstrate this endpoint in a clinical trial, although evidence of granulomatous inflammation should be a requirement for study entry.
Because corticosteroids are highly efficacious for pulmonary sarcoidosis, additional anti-granulomatous agents are often used as corticosteroid-sparing agents in order to lower the corticosteroid dose requirement. Additional agents can also be used to provide further anti-granulomatous therapy on top of corticosteroids, although often the additional benefit of such agents is minimal in pulmonary sarcoidosis patients receiving more than 10 to 15 mg of prednisone per day60. Figure 5 outlines how a corticosteroid-sparing agent can effectively decrease the corticosteroid dose sweet spot in terms of the benefits of corticosteroid therapy versus the risks of corticosteroid toxicity. A majority of 36 sarcoidosis experts believed that a chronic daily maintenance dose of more than 10 mg of prednisone was unacceptable and mandated consideration of corticosteroid-sparing therapy61. For pulmonary sarcoidosis, these corticosteroid-sparing agents include anti-metabolites such as methotrexate62,63, azathioprine64,65, and the tumor necrosis factor-alpha (TNF-α) antagonists infliximab56 and adalimumab66. These agents usually are used as second- or third-line agents that are added on to corticosteroid therapy because they either take weeks to months longer than corticosteroids to be effective63,65 or—in the case of the TNF-α antagonists—are much more expensive than corticosteroids.
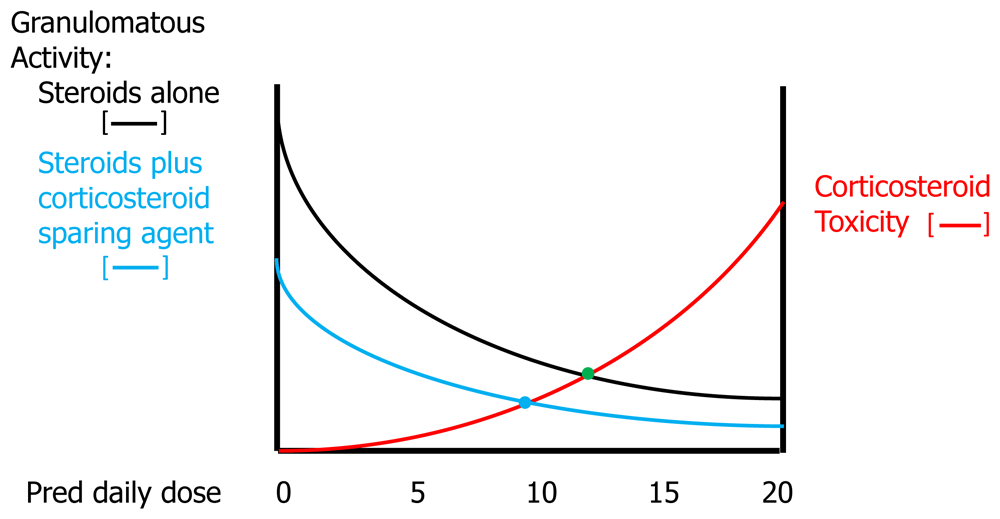
A corticosteroid-sparing drug has some anti-granulomatous activity such that a lower dose of corticosteroids (blue line) is needed to achieve the same level of granulomatous activity as without use of the corticosteroid-sparing medication (black line). This results in lowering the sweet spot for the ideal corticosteroid dose (from the green dot to the blue dot).
Entry criteria for anti-granulomatous therapy trials for patients with chronically treated pulmonary sarcoidosis should include evidence of granulomatous inflammation, physiologic impairment, and functional/QOL impairment at study entry or—if a corticosteroid withdrawal phase of the trial is planned—historical evidence of these abnormalities when the corticosteroid dose is lowered. Figure 6 outlines the effect on pulmonary physiology with the addition of a corticosteroid-sparing anti-granulomatous agent for chronically treated pulmonary sarcoidosis. Because corticosteroids are effective anti-granulomatous agents, there is often relatively little residual granulomatous inflammation that requires additional therapy. The prototypical chronic pulmonary sarcoidosis patient enrolled in a clinical trial is receiving chronic corticosteroid therapy. The patient’s pulmonary function has usually waxed and waned over time, depending on the dose of corticosteroids and other anti-granulomatous drug therapy. The pulmonary function of these patients may decline if their granulomatous inflammation is undertreated, and they may develop permanent reductions in pulmonary function from the development of pulmonary fibrosis. However, this fibrosis rarely causes a rapid decline in physiology, as evidenced by the fact that the placebo group in several trials where all subjects were receiving a maintenance corticosteroid medication in addition to a study drug did not demonstrate a significant reduction in spirometry over a study period of 4 months or more55,56.

There is usually minimal residual granulomatous inflammation in patients who receive corticosteroids such that there is a small potential for further significant physiologic improvement (green double arrow). It is important to note that although additional drug therapy may not greatly improve physiology, it may significantly improve quality of life by reducing corticosteroid side effects if addition drugs are corticosteroid-sparing.
For these reasons, the endpoints of trials for acute untreated pulmonary sarcoidosis are not highly relevant to evaluate anti-granulomatous drugs used in chronically treated pulmonary sarcoidosis patients. There is often little granulomatous inflammation left to treat, and the issue is whether the drug will continue to suppress granulomatous inflammation when the corticosteroid dose is lowered. The same could be said for the physiologic endpoint: the issue is often not whether the additional drug will improve pulmonary physiology but rather whether it will allow the pulmonary physiology to be maintained as the corticosteroid dose is lowered. The QOL endpoint is still highly relevant, especially in terms of a reduction of corticosteroid side effects. Table 4 outlines potential endpoints for a trial of anti-granulomatous therapy for chronically treated pulmonary sarcoidosis.
As previously mentioned, it is conjectured that the fibrosis in sarcoidosis is the result of granulomatous inflammation. The relationship of granulomatous inflammation, pulmonary fibrosis, and treatment of pulmonary sarcoidosis is depicted in Figure 7. The amount of fibrosis that develops with pulmonary sarcoidosis is highly variable and is essentially unpredictable in individual patients (Figure 7A). The physiologic defects that develop from fibrosis in pulmonary sarcoidosis are permanent and will not respond to anti-granulomatous therapy or any other therapies that are currently available (Figure 7B).
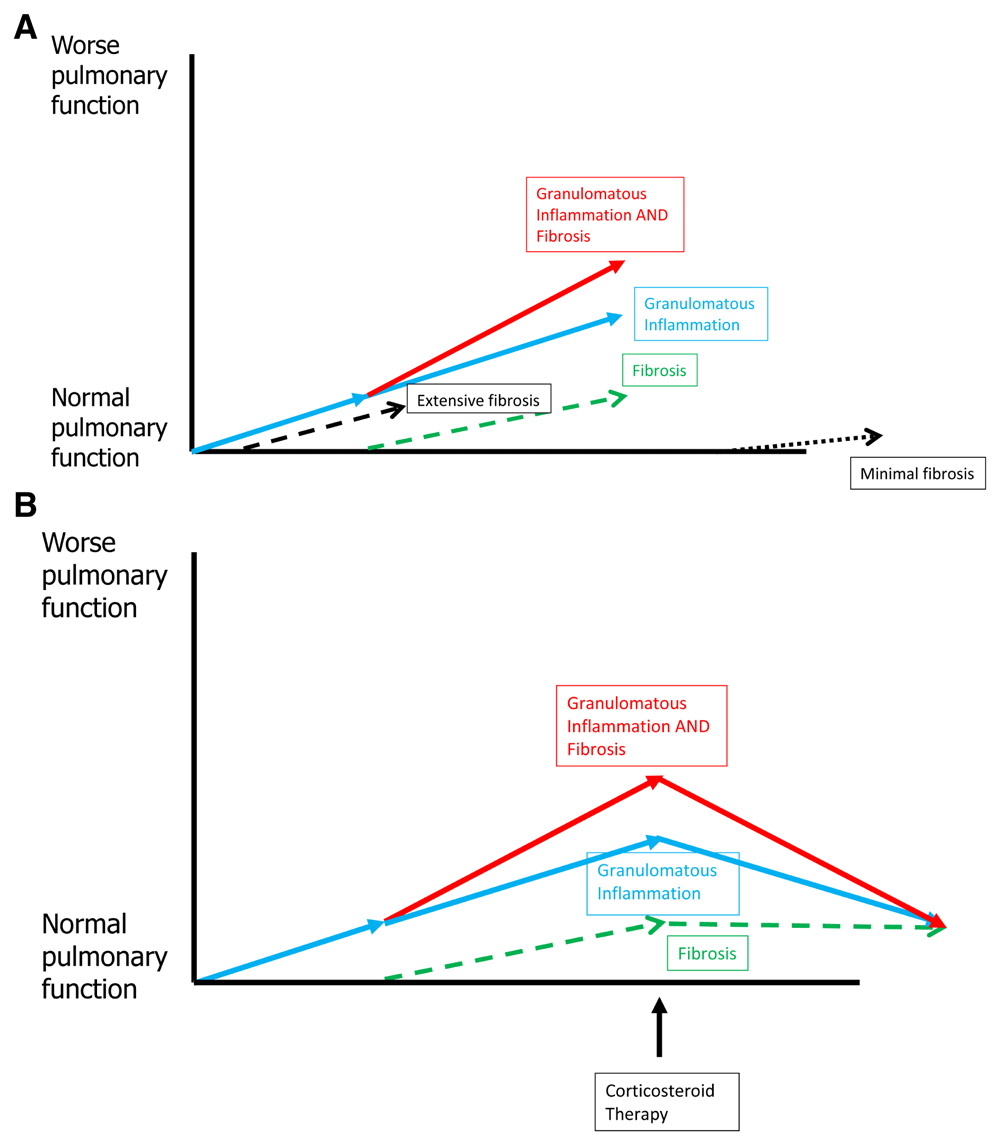
(A) Development and progression of pulmonary fibrosis in a patient with active pulmonary sarcoidosis. The granulomatous inflammation of sarcoidosis may result in worsening pulmonary symptoms and worsening pulmonary function (blue line). Because the granulomatous inflammation of sarcoidosis is the most common cause of pulmonary fibrosis in sarcoidosis, fibrosis may develop as the granulomatous inflammation continues (green line). This fibrosis contributes to worsening pulmonary symptoms and worsening pulmonary dysfunction (red line). In some patients with pulmonary sarcoidosis, the fibrotic reaction to the granulomatous inflammation may be robust (wide dotted line), causing extensive fibrosis early. Other patients with pulmonary sarcoidosis may have a sluggish fibrotic response to the granulomatous inflammation (short dotted line), and the fibrosis is minimal and requires granulomatous inflammation to be active for a longer period of time. Some patients with pulmonary sarcoidosis may not develop fibrosis at all in response to the granulomatous inflammation. (B) The effect of treatment on the granulomatous inflammation and fibrosis associated with pulmonary sarcoidosis. Effective anti-granulomatous therapy will suppress the granulomatous inflammation of sarcoidosis (blue line) and improve the overall pulmonary symptoms and pulmonary function (red line). However, anti-granulomatous therapy will have no effect on the pulmonary symptoms or dysfunction caused by sarcoidosis-related fibrosis (green line). Therefore, successfully treated patients will have their symptomatic and physiologic improvement limited by the degree of pulmonary fibrosis that has developed.
Therapy for fibrotic pulmonary sarcoidosis is currently aimed at either (a) treating the complications of pulmonary fibrosis or (b) preventing the progression of further lung fibrosis. The complications of pulmonary fibrosis are listed in Table 1 and include sarcoidosis-associated pulmonary hypertension67,68, bronchiectasis69,70, and pulmonary mycetoma71,72. The indications for treatment of these conditions as well as the endpoints to monitor these forms of disease are well covered elsewhere69,71,73–75. Anti-granulomatous therapy is rarely useful for sarcoidosis-associated pulmonary hypertension and is often detrimental for bronchiectasis and pulmonary mycetoma as it may cause serious infectious complications. As end-stage pulmonary fibrosis is a disabling and life-threatening condition, lung transplantation should be considered in fibrotic pulmonary sarcoidosis patients with limited chance for improvement and minimal extrapulmonary comorbidities76.
In terms of therapy to prevent the development of further fibrosis, it has already been mentioned that the majority of patients with fibrotic pulmonary sarcoidosis have evidence of granulomatous inflammation16,19. It is thought that such “smoldering” granulomatous inflammation is the nidus for the development of further fibrosis. Therefore, evidence of granulomatous inflammation would be a prerequisite for instituting anti-granulomatous therapy for fibrotic pulmonary sarcoidosis.
An additional issue when considering treatment of sarcoidosis-associated pulmonary fibrosis is the rapidity of its development. It is thought that fibrosis develops slowly in pulmonary sarcoidosis, such that the cumulative toxicities of long-term anti-sarcoidosis medications, especially corticosteroids, would limit their long-term use. Certainly, evidence of rapid development of pulmonary fibrosis with a concomitant rapid decline in physiology or functional status/QOL would be a strong impetus to consider anti-granulomatous therapy. Because of the toxicity of corticosteroids and other anti-granulomatous therapies, another approach would be to use “downstream” medications targeting the development of fibrosis itself. Such medications include pirfenidone and nintedanib, which have been shown to decelerate the development of lung fibrosis in idiopathic pulmonary fibrosis and other interstitial lung diseases77–79.
In terms of endpoints for the treatment of fibrotic pulmonary sarcoidosis, improvements in pulmonary function and functional status/QOL from anti-granulomatous therapy may be significant yet are unlikely to be large because improvement in the patient’s pulmonary function is unlikely to be great57. The ablation of granulomatous inflammation may improve QOL by reducing symptoms derived from parasarcoidosis syndromes that may be indirectly derived from granulomatous inflammation (see below). However, other useful endpoints for the treatment of fibrotic pulmonary sarcoidosis may reflect a lack of deterioration of patient status. Table 5 shows potential indications for the treatment of fibrotic pulmonary sarcoidosis, and Table 6 shows potential endpoints of treatment for fibrotic pulmonary sarcoidosis.
| Indication for treatment | Rationale | Negative features |
|---|---|---|
| The presence of granulomatous inflammation | Treating active granulomatous inflammation in fibrotic pulmonary sarcoidosis improves physiology | Fibrosis in sarcoidosis usually develops slowly such that obliteration of granulomatous inflammation may have a minimal appreciable effect on the development of fibrosis. |
| Significant impairment in functional status or quality of life from previous pulmonary fibrosis | Usually the major concern of patients and clinicians | No anti-fibrotic therapy that lessens the degree of pulmonary fibrosis is currently available. |
| Relatively rapid decline in functional status or quality of life impairment from pulmonary fibrosis | Usually the major concern of patients and clinicians | No anti-fibrotic therapy for pulmonary sarcoidosis is currently available. |
| Development of a condition associated with fibrotic pulmonary sarcoidosis (Table 1) | These conditions may cause significant impairment of quality of life or dangerous conditions or both | Most of these treatments are for complications of pulmonary fibrosis and do not directly treat the presence or worsening of pulmonary fibrosis. |
The physiologic impairment resulting from the granulomatous inflammation of pulmonary sarcoidosis is highly variable. Even more variable is the effect of these physiologic derangements on the QOL of the patient with pulmonary sarcoidosis. This granulomatous inflammation can also adversely affect the QOL of the patient with sarcoidosis by systemic mechanisms other than direct deposition of granulomas into specific organs (“parasarcoidosis syndromes”). Additionally, this granulomatous inflammation may lead to the development of pulmonary fibrosis that causes irreversible pulmonary dysfunction and, potentially, disabling and life-threatening situations. Finally, anti-granulomatous therapy not only may improve the QOL of the patient with pulmonary sarcoidosis by effectively treating the disease but also may worsen it by causing serious drug side effects. We have proposed a conceptual framework to outline how these many factors interact to cause physiologic and QOL impairments in pulmonary sarcoidosis. We believe that this conceptual framework may be useful in constructing a clinical drug trial for pulmonary sarcoidosis that reflects the concerns of patients and clinicians.
Because of numerous aspects of pulmonary sarcoidosis and its therapy, it is not surprising that developing treatment indications and endpoints for the therapy of the disease is challenging. We have made a case for partitioning patients into three treatment groups when considering indications for therapy as well as therapy endpoints: untreated patients with active granulomatous inflammation, patients with chronically treated disease, and those with significant lung fibrosis. It is hoped that this proposed patient partitioning will stimulate debate and constructive criticism that will eventually lead to therapy for pulmonary sarcoidosis that is better directed in treating the true concerns of patients.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)