Keywords
Pseudomonas aeruginosa, transcriptional regulator, LasR, antibiotic resistant, ESKAPE, 3OC12-HSL, homology modelling, molecular docking, molecular dynamics, machine learning techniques
Pseudomonas aeruginosa, transcriptional regulator, LasR, antibiotic resistant, ESKAPE, 3OC12-HSL, homology modelling, molecular docking, molecular dynamics, machine learning techniques
PDB, Protein Data Bank; MD, molecular dynamics; PCA, principal component analysis; 3OC12-HSL, N-3-oxododecanoyl homoserine lactone; AI, autoinducer; SLR, short linker region; BLAST, Basic local alignment search tool; DBI, David-Bouldin index; pSF, pseudo-F statistic; IQS, integrated quorum sensing; QS, quorum sensing; AHL, acyl homoserine lactone; HCN, hydrogen cyanide; HTH, helix-turn-helix; MSA, multiple sequence alignment; RMSD, root-mean-square-deviation; LBD, ligand binding domain; DBD, DNA binding domain
Pseudomonas aeruginosa (P. aeruginosa) is a Gram-negative, monoflagellated, obligate aerobe1,2. This species of bacteria is one of the ‘ESKAPE’ pathogens, which includes six bacterial pathogens which are commonly associated with antimicrobial resistance and are the leading cause of nosocomial infections throughout the world3. It can be found in many diverse environments such as in soil, plants and hospitals1,4. P. aeruginosa is an opportunistic human pathogen because it rarely infects healthy people, mostly affecting patients suffering from AIDS, cystic fibrosis, cancer and burns2,5. Most of the deaths caused by cystic fibrosis are due to this pathogen1,6. The pathogenicity of P. aeruginosa is due to virulence factors such as: the synthesis of proteases, hemolysins, exotoxin A, pyocyanin, hydrogen cyanide (HCN) and rhamnolipids; the possession secretion systems types one (T1SS), two (T2SS), three (T3SS), four (T4SS)7, five (T5SS), and six (T6SS)8; the formation of biofilms7,9.
Biofilm formation is a common characteristic of many bacteria as the bacterial population density increases in the body during pathology. These bacteria possess a system of regulating gene expression in response to population density, called quorum sensing (QS), that uses hormone-like molecules called autoinducers (AIs) which accumulate in the extracellular matrix. When a threshold is reached, the AIs bind to their cognate receptors and then a response regulator modulates the expression of QS virulence genes which regulate adaptation, colonization, antibiotic resistance, plasmid conjugation and other bacterial processes.
Pseudomonas aeruginosa has four QS systems10. The type one system is regulated by LuxI/LuxR-type proteins. AIs diffuse freely across the bacterial membrane and bind to the transcriptional activator LuxR. Type two and three systems are also LuxI/LuxR-type systems, the first LasI that produces 3OC12-HSL and the second, RhlI that synthesizes C4-HSL; both acyl homoserine lactones (AHL) which regulate virulence and biofilm formation. A fourth integrated QS (IQS) system has been characterized very recently and the genes involved in IQS synthesis are non-ribosomal peptide synthase genes of the ambBCDE operon. The transcriptional regulator of IqsR is the IQS receptor11.
QS regulates the expression of virulence factors such as HCN. Exposure to HCN can lead to neuronal necrosis through the inhibition of cytochrome c oxidase, the terminal component of the aerobic respiratory chain12,13. In P. aeruginosa, the hcnABC operon is responsible for HCN biosynthesis by the enzyme HCN synthase14. Three transcriptional regulators (LasR, ANR and Rh1R15) control the transcription of the hcnABC gene cluster14 although it has been proposed that LasR is the crucial activator of hcnABC genes following mutagenesis experiments16.
The transcriptional activator protein LasR regulates target gene expression by recognizing a conserved DNA sequence termed a lux box16,17. LasR has two domains: 1) a ligand binding domain at the N-terminus (LBD); 2) a DNA-binding domain at the C-terminus (DBD)18. LasR has a DNA-binding helix-turn-helix (HTH) motif19. Binding of the AI 3-O-C12HSL stabilizes LasR and promotes its dimerization. After that, the resulting LasR-AI homodimer complex binds to the promoter of the target DNA and induces gene transcription18.
During colonization and invasion, both the pathogen and the host are exposed to molecules released by each other, such as bacterial AIs or host stress hormones and cytokines. The mechanisms and receptors that might be involved in cross-talk between microbial pathogens and their hosts are not yet well described. LuxR homologue studies have demonstrated that LuxR-type activators are homodimers and consist of two domains. These two functional domains are joined by a short linker region20,21.
There remains a need for a further understanding of the LasR monomer because to date there is no detailed information about LasR monomer interactions. Hence, we analysed the molecular details of the interactions of 3OC12-HSL with the LasR protein. So far, this is the first report that shows that 3OC12-HSL can interact with conserved amino acid residues from the ligand binding domain (LBD), beta turns in the short linker region (SLR) and the DNA-binding domain of LasR. For simplification, we refer to this region as the LBD-SLR-DBD bridge or “the bridge”. This could mean that modulation of the transcriptional regulator LasR is more nuanced than previously thought. This study may be utilized for the development of new therapeutic agents against P. aeruginosa targeting both the LBD and the LBD-SLR-DBD bridge of LasR in order to inhibit the expression of virulence genes.
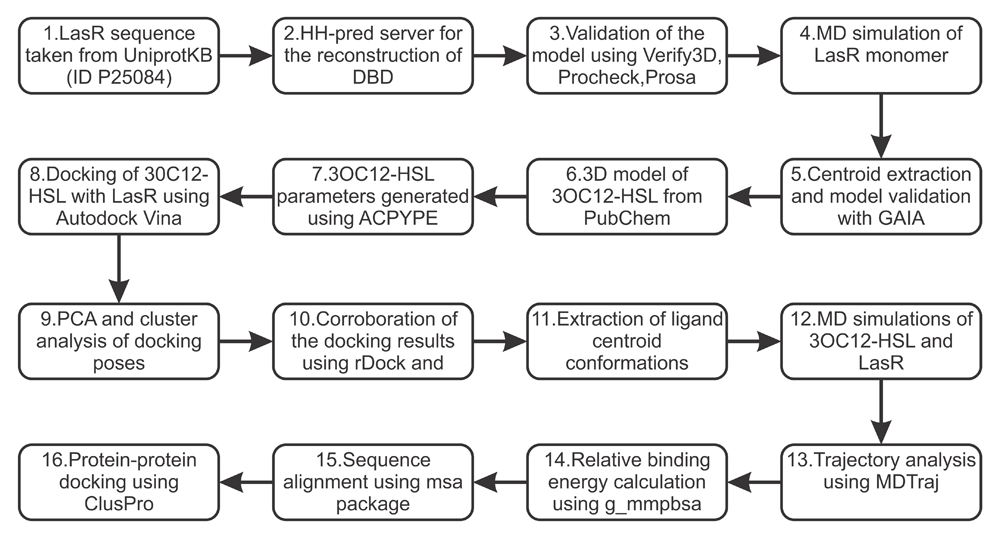
Figure 1. Flow chart providing an overview of the methodology used in this study.
This part of the methodology was based on the work by Chowdhury et al. to reproduce the full structure of LasR protein1. The amino acid sequence of the LasR protein of P. aeruginosa was obtained from UniprotKB22 (Uniprot ID: P25084). The crystal structure of the LasR protein LBD (amino acid residues 7 to 169) from P. aeruginosa was acquired from the Protein Data Bank23 (PDB ID: 3IX3). However, the entire structure of the LasR protein was required in order to have a better understanding of its molecular properties.
For this reason, the amino acid sequence (Uniprot ID: P25084) was used to search the PDB databank using BLASTp24 in order to identify suitable templates for homology modelling. The crystal structure of QscR bound to 3OC12-HSL was found to be the best fit based on sequence identity, query coverage, and E-value from the BLAST results (PDB ID: 3SZT)20. The full list of similar structures obtained from the BLAST search are shown in Table 1.
As the current available crystal structures of LasR only contain the ligand binding domain18, we needed to reconstruct the full structure of LasR with the missing DNA-binding domain (DBD). The HHPred web server25 was used for homology modelling of the missing LasR DBD as was performed by Chowdhury et al.1. The four templates used for homology modelling of the LasR DBD have the PDB identifiers 3SZT, 3IX3, 1FSE and 3ULQ. The experimental structure of LasR18 and these templates were used to reconstruct the final model of LasR that contains both the LBD and the DBD. The final reconstructed model of the LasR protein was verified using PROCHECK25 (Figures S1 and S2, Extended data26), Verify3D26 and Ramachandran plots. More than 96.65% of the amino acid residues in the final reconstructed model had a 3D–2D score > 0.2 as indicated by Verify3D (Figure S3, see Extended data26). This score indicates a suitable computational model27. The Ramachandran plot (Figure S1, see Extended data26) revealed that no amino acid residues were located in the disallowed regions. The ProSA28 value of this model was -6.69 which suggests that the quality of the homology model is high28. Although the obtained model is slightly different from that obtained by Chowdhury et al.1 it is still viable.
The analysis of docking conformations and trajectories was performed using custom Python scripts (see Extended data26). These scripts use pandas v0.8.129, scikit-learn v0.18.230 and MDTraj v1.931 libraries. pandas29 facilitates data manipulation and analysis. In particular, it offers data structures and operations for manipulating numerical tables and time series. scikit-learn30 is a machine-learning library, which provides a set of common algorithms to Python users through a consistent interface. The MDTraj Python library31 facilitates the combination of machine learning libraries such as scikit-learn for trajectory analysis. MDTraj serves as a bridge, connecting molecular dynamics (MD) data with statistics and graphics libraries developed for general data science users.
Diagrams of the interaction of 3OC12-HSL with LasR were obtained using LigPlot+ v.1.432 using default parameters, which automatically generates 2D diagrams of ligand–protein interactions from 3D coordinates by loading the coordinate files in pdb format. Plot visualization was carried out using matplotlib v2.0.132,33 with seaborn v0.8.134. The seaborn library aims to make visualization a central part of exploring and understanding data. It also provides a concise, high-level interface for drawing statistical graphics. Figures and videos were prepared with PyMOL v1.9.0.035, VMD 1.9.336 and UCSF Chimera v1.11.237. VMD36 is often used for this purpose as it has the ability to read trajectory files created during simulations in formats produced by many different software packages. PyMOL35 and UCSF Chimera37 are also commonly used by researchers and produce high quality images but are typically less straightforward to use for viewing and making videos of the trajectories.
The analysis protocol was similar to the work of Wolf et al.38 and Zamora et al.39, using PCA and cluster analysis modules within scikit-learn v0.18.2. Principal component analysis (PCA) is an unsupervised statistical technique that is often used to make data easy to explore and visualize. PCA tries to explain the maximum amount of variance with the fewest number of principal components40. The process of applying PCA to a protein trajectory is called essential dynamics (ED)41,42. PCA analysis, performed using Cartesian coordinates, has become an important tool for the examination of conformational changes. Default parameters within the scikit-learn library were used. Cluster analysis is another unsupervised technique that tries to identify structures within data. It is a data exploration tool for dividing a multivariate dataset into groups. Clustering algorithms can be grouped into partitional and hierarchical clustering approaches43. kmeans algorithm44,45 was used for molecular docking analysis. Agglomerative clustering using Ward’s linkage46 was used for molecule dynamics trajectory analysis with the connectivity matrix built on the basis of the k-Neighbors graph30. Default parameters within the scikit-learn library were used.
Molecular dynamics (MD) simulations of all systems were conducted with the GROMACS suite, version 5.1.247, utilizing the Amber ff99SB-ILDN force field48, which exhibits considerably better agreement with NMR data than other forcefields. In all cases, short-range non-bonded interactions were cut off at 1.4 nm. The Particle mesh Ewald (PME) method49,50 was used for the calculation of long-range electrostatics.
LasR monomer simulation. In order to generate the starting structure of the LasR monomer before docking, the Las R molecule was placed in a dodecahedron box of TIP3P water51. After that, 100 mM NaCl was added, including neutralizing counterions. Following two minimizations by the method of steepest descent algorithm, the LasR monomer was equilibrated in two stages. The first stage involved simulating for 200 picoseconds under a constant volume (NVT) ensemble. The second stage involved simulating for 200 picoseconds under a constant pressure (NpT), maintaining pressure isotropically at 1.0 bar. Production simulation was conducted for 100 nanoseconds in the absence of any restraints. The temperature was sustained at 300 K using the v-rescale52 algorithm. For isotropic regulation of the pressure, the Parrinello-Rahman barostat53 was used. This combination of thermostat and barostat use ensures that an adequate NpT ensemble is sampled. Finally, the trajectory of the LasR monomer simulation was used for the calculation of chemical shifts. SPARTA+ v2.9054 and SHIFTX2 v1.1055 were used to predict the chemical shifts of the protein backbone atoms with the help of the MDTraj wrapper functions31. Both programs have been proven to be remarkably accurate, especially for predicting 13C shifts54. This was used to validate whether the force field is appropriate for further investigation. The final extracted structure that was used for docking and MD simulations was also verified with Gaia56 from the Chiron webserver57, which compares the intrinsic structural properties of in silico protein models to high resolution crystal structures.
Molecular dynamics simulations using docking poses. The 3D model of 3OC12-HSL was obtained from PubChem (CID: 127864) (Figure 2)58. It has been shown that docking has its limitations59. For this reason, after finishing the molecular docking simulations of 3OC12-HSL with LasR, the docking poses were extracted to perform molecular dynamics (MD) simulations. The ligand parameters for the molecular dynamics simulations were calculated for the General Amber Force Field (GAFF)60 using the ACPYPE tool61 with AM1-BCC partial charges62 and generation of a GAFF topology. A time step of 2 femtoseconds was used during heating and the production run and coordinates were recorded every 1 picosecond. Three simulations of 300 nanoseconds were performed. Further details about the simulation protocol can be found in ‘LasR monomer simulation’ section.
To build the LasR–ligand complex, the ligand 3OC12-HSL was docked with the LasR monomer using AutoDock Vina v1.1.263. However, AutoDock Vina currently uses several simplifications that affect the obtained results. The most notable simplification, as the creators note63, is the use of a rigid receptor. The amount of computation used during a docking experiment is determined by a parameter called ‘exhaustiveness’ in AutoDock Vina63,64. However, the default exhaustiveness value is 8 and the creators claim that increasing this value leads to an exponential increase in the probability of finding the energy minima63,64. The conformational space of the whole protein was searched using grid box dimensions 60×62×48Å. The following exhaustiveness values were tested in this study: 8, 16, 32, 64, 128, 256, 512, 1024, 2048 and 4096. The ligand and target structures containing hydrogen atoms were prepared using the AutoDockTools65 from MGLTools v1.5.6.
Principal component analysis (PCA)40 and cluster analysis using k-means clustering44,45 was performed (Figures S4 and S5, Extended data26). The results demonstrated that the number of interaction sites does not change for exhaustiveness values from 1024 to 4096. An exhaustiveness value of 1024 was chosen as it provides good results, good speed and thorough sampling of the docked configurations. The exhaustiveness value was set to 1024 and the maximum number of binding modes to generate was set to 20. After that, 100 independent docking calculations were carried out with random initial seeds. The target was taken from the molecular dynamics simulation of the LasR monomer. Water molecules were removed from the structure.
We also performed another set of docking simulations using AutoDock Vina v1.1.263, rDock v2013.166 and FlexAID v2.4867. We performed 10 docking simulations using each program. Each individual run was set to generate 20 docked poses. For AutoDock Vina, we performed one docking run for each exhaustiveness value: 8, 16, 32, 64, 128, 256, 512, 1024, 2048 and 4096.
Docking was also performed using the rDock program66 which was previously found to be successful in predicting the binding mode for the CCDC-Astex set of 85 diverse protein–ligand complexes in approximately 80% of cases66. A cavity with a radius of 36 Å was set and centred on the LasR monomer to cover the whole protein surface and define the docking volume. AutoDock Vina and rDock were chosen based on the performance of these academic programs68.
FlexAID (parameters are available in Extended data26) was also used as, unlike other programs, it is robust against increasing structural variability67. FlexAID performs better than AutoDock Vina in all scenarios against non-native complex structures63. FlexAID uses a soft and smooth scoring function based on contact surfaces which is different from Autodock Vina and rDock. FlexAID was run using default parameters.
g_mmpbsa v1.669 was used for the evaluation of binding energies. It was also used for the estimation of the energy contribution of each residue to the binding energy. g_mmpbsa calculates the relative binding energy rather than the absolute binding energy, so the entropy contribution (T∆S) is not evaluated69. From the two simulations with length of 300 nanoseconds, we cut out 10 nanoseconds with an interval of every 120 picoseconds for the calculation of binding energies. g_mmpbsa uses the MM-PBSA approach, which has grown to be one of the most widely used methods to compute interaction energies and is often employed to study biomolecular complexes65.
To find out whether 3OC12-HSLs interact with conserved amino acid residues, we performed multiple sequence alignment (MSA) using the R msa package v1.4.570. ClustalW71, Clustal Omega72 and MUSCLE73 within the msa package were used to carry out multiple sequence alignments. ClustalW and MUSCLE are commonly and widely used, while Clustal Omega is a more recent and powerful method that is used when aligning a large number of sequences72. We performed multiple sequence alignment of the LasR protein and closest homologue sequences, based on the work by Chowdhury et al.1. The consensus threshold parameter in msa was set to 75, the rest was set to default. Table 2 shows the sequences used for sequence alignment.
When docking homology models, it is preferable to have experimental evidence suggesting the general interaction site (within ~10 Å). Representative structures from molecular dynamics simulations were used for protein-protein docking using ClusPro v2.074. ClusPro was chosen because it gave equivalent results to the best human predictor group according to the latest CAPRI experiments carried out in 201368.
From the experimental X-ray data, it was found that ‘Trp152’, ‘Lys155’ and ‘Asp156’ play an important role during dimerization. The distance between ‘Trp152’ from chain A and ‘Asp156’ from chain B of the crystallographic structure was 0.276 nm and the distance between ‘Asp156’ from chain A and ‘Lys155’ from chain B was 0.279nm. These residues were used as attraction constraints.
We performed a simulation run of 100 nanoseconds using a standard MD protocol for the assessment of conformational changes of the LasR monomer. The overall stability of the molecule was assessed using the root-mean-square-deviation (RMSD) of the protein atoms. RMSD was calculated with reference to the initial frame through the time of the MD run (Figure S6, Extended data26). Another suitable measurement for the stability of the LasR monomer structure is the radius of gyration, which also shows the structure is stable (Figure S7, Extended data26).
During the examination of MD trajectories, principal component analysis (PCA)40 is usually used for the visualization of the motion of the system (Figure 3a). Generally, in order to capture more than 70% of the variance in the trajectory data, the first handful of components are sufficient39. PCA can uncover the fundamental movements contained in an MD trajectory, however, it does not group the snapshots into different clusters43. This can be accomplished by clusterization of the PCA data.
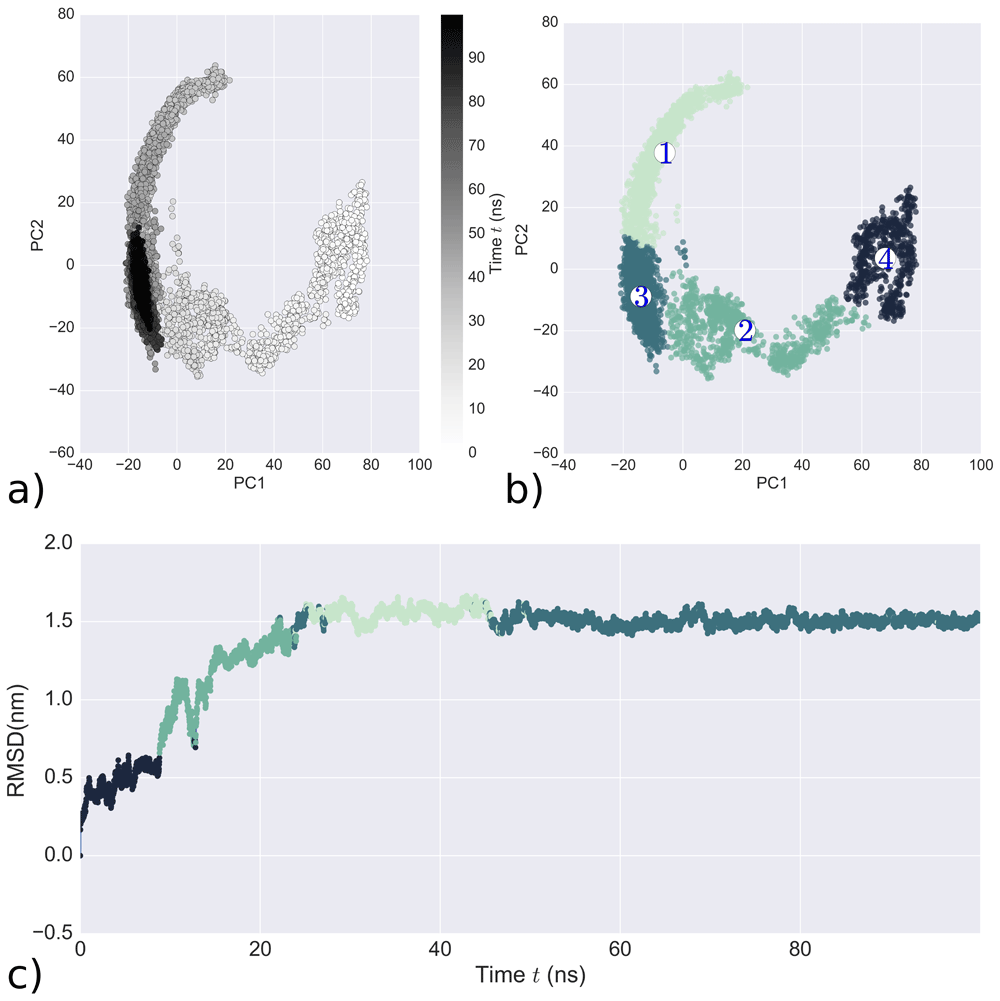
LasR monomer simulation analysis a) Simulation trajectory projected onto its first two principal components. The black scale indicates the time progression from 0 ns (white) to 100 ns (black) b) Clustering results of ward-linkage algorithm formed by first two PCs c) Colour-coded RMSD of the simulation obtained from Ward-linkage cluster analysis.
Cluster analysis was performed on the LasR monomer simulation for the selection of an initial starting structure to study docking. Identifying a distinct, representative structure of the cluster allows blind docking to be performed on the whole structure. It should be also noted that cluster analysis allows the evaluation of the frequent conformations of LasR. For the clustering analysis, we chose agglomerative clustering using Ward’s linkage46. This algorithm appears to be the most appropriate as it is deterministic, allowing for reproducibility of the resulting clusters, thus minimizing the amount of bias.
We performed several rounds of agglomerative clustering using Ward’s linkage. The accuracy of the clustering was assessed with the help of the Davies–Bouldin index (DBI)75, Dunn index76, silhouette score77 and Calinski-Harabasz pseudo-F statistic (pSF)78 metrics. An optimal number of clusters were chosen, simultaneously accounting for low DBI, high silhouette, high Dunn index and high pSF values.
The distribution of clusters over the simulation is visualized in Figure 3b and the four clusters are: cluster 4 (black) at the beginning of the simulation (after equilibration), cluster 2 (green) in the middle, cluster 1 (light green) and cluster 3 (dark blue) at the end. The clusterization defined by the first two principal components (Figure 3b) provides a coherent picture and it is also supported by good DBI, Dunn index, silhouette score and pSF values (Figure S8, Extended data26). Figure 3c shows clearly that the simulation has converged.
For validation of the molecular dynamics simulation quality, theoretical NMR shifts were calculated using Sparta+51 and ShiftX252. For the NMR shifts calculation, snapshots from cluster 3 (Figure 3b) were used because of their stability. We compared NMR values of C-alpha atoms from the experimental NMR of the LBD with theoretical NMR values of the LBD from our molecular dynamics simulations (BMRB: 6271)18. Unfortunately, the current available NMR information only includes the 173 amino acid residues of the LBD. In Figure 4, one can see that simulated NMR shift values from the two programs are close to the experimental data, thus demonstrating the quality of the MD simulation of the LasR monomer.
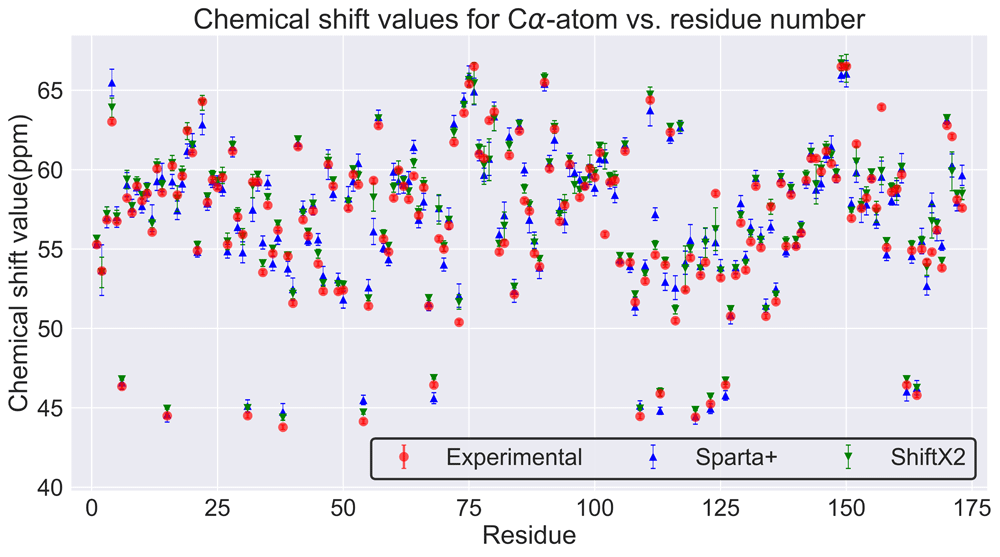
It should be noted this protein becomes partially soluble when the ligand interacts with it18. The differences between the theoretical chemical shifts and experimental values could be attributed to: 1) the theoretical model does not involve the interaction with the ligand 2) the experimental NMR only includes information about the LBD and not the DBD, which can affect the LBD.
After that, the representative structure (Figure 5c) was extracted from cluster 3 (Figure 3c) to study docking. The representative structure of the LasR system and its differences from the homology modelled structure (Figure 5b) are visible in Figure 5c. Secondary structure analysis of the representative structure was performed using PDBSum79 (Figure S9, Extended data26). Finally, the extracted centroid structure was validated using Gaia56 (Figure S10, Extended data26), which also shows the model is of a high quality. Therefore, in this study, a model has been developed to include the dynamics of the full-length LasR molecule (residues 1 to 239) and it has been shown that the dynamics of the complete C-terminal region of LasR modulate the N-terminal region, as will be discussed later.
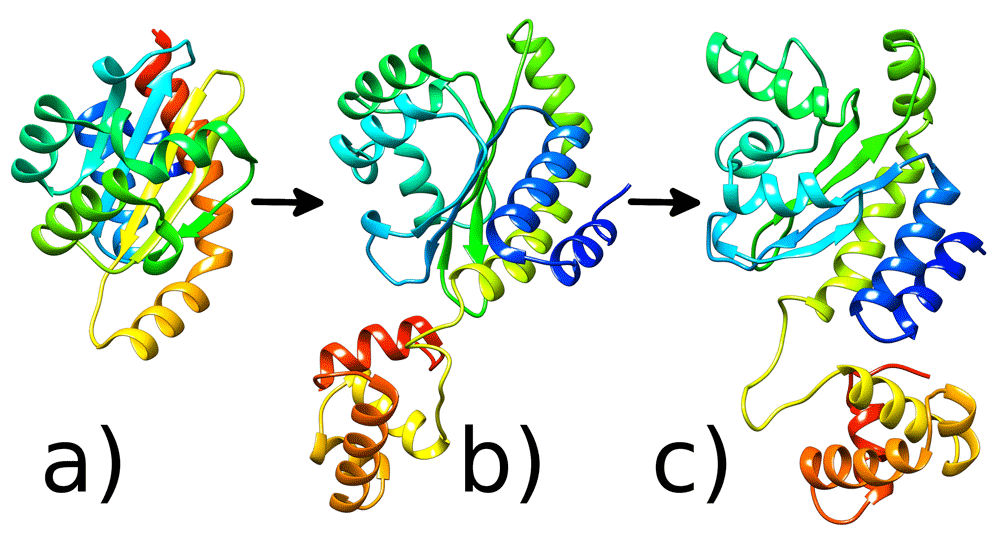
3D LasR protein structures: a) Crystal structure of the N-terminal LBD of LasR protein b) Full structure of the protein with the missing DBD, which was modelled using homology modelling c) Representative structure after 100ns MD run. Images generated with UCSF Chimera36.
In this study, a molecular docking approach was used to inspect the possible binding modes of 3OC12-HSL with the LasR monomer. PCA and cluster analysis were performed on docking data (Figure 6a, b) and the results show three binding sites. Cluster 2 corresponds to experimental data, while cluster 1 and cluster 3 do not (Figure 6c). These results clearly support the findings of Bottomley et al.18, who also demonstrated that 3OC12-HSL binds to the LBD.
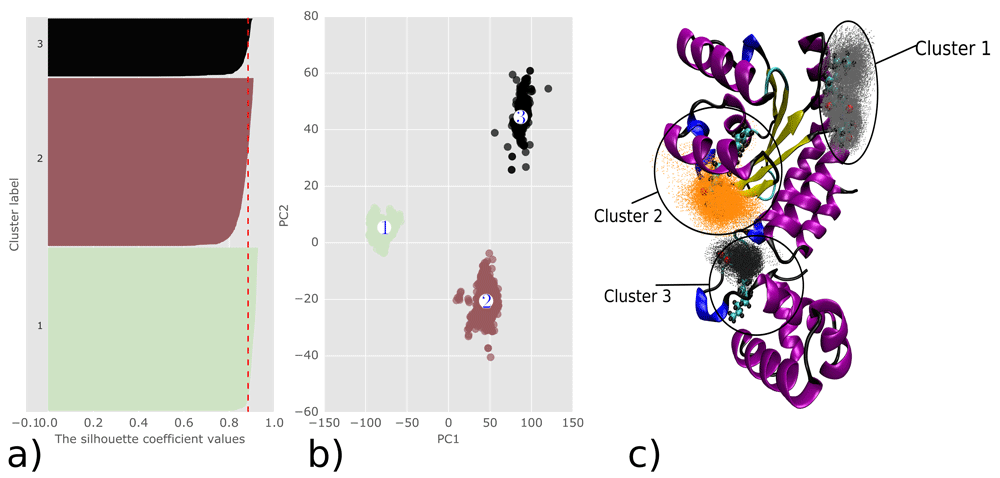
Analysis of Autodock Vina docking results a) Silhouette plot analysis of clusterization quality b) Clustering results using k-means algorithm formed by first two PCs c) 3D visualization of the analysed docking data with their representative structures and clusters.
We performed several rounds of k-means clustering. The accuracy of the cluster analysis was evaluated using the DBI75, Dunn index76, silhouette score77 and pSF78 metrics (Figure S11, see Extended data26). An optimal number of clusters were chosen for docking analysis, simultaneously accounting for low DBI, high Dunn, high silhouette and high pSF values. We generated 2000 docked poses and performed representative structure extraction for use in MD simulations of the LasR 3OC12-HSL binding sites. The representative structures were produced by identifying the centroid conformations of the clusters using the following algorithm:
1. Compute all pairwise root mean square deviations (RMSDs) between the conformations.
2. Transform the distances into similarity scores and they are calculated as
Where sij is the pairwise similarity, dij is the pairwise distance, and dscale is the standard deviation of the values of d to make the computation scale invariant.
Representative structures from each cluster were extracted for further use in MD simulations. The binding energy of the representative structure of cluster 1 is -5.4 kcal/mol and the mean binding affinity for the whole cluster is -5.257 ±0.233 kcal/mol. Cluster 1 contains 839 docked poses from a total 2000 (41.95%). For cluster 2, the binding affinity of the representative structure is -5.1 kcal/mol and the mean for the whole cluster is -5.593 ±0.386 kcal/mol. These results correspond to the experimental binding site data18. Cluster 2 contains 864 docked poses from a total of 2000 (43.2%). For cluster 3, the representative structure features the highest binding affinity of -5.7 kcal/mol and the mean binding affinity for the whole cluster is -5.264 ±0.27 kcal/mol. Cluster 3 contains 297 docked poses from 2000 (14.85%) which does not correspond to the experimental binding site.
It has been suggested that molecular docking is not appropriate for the prediction of binding affinity or binding poses of protein-ligand complexes. However, docking analysis can still provide important information regarding potential binding sites and the resulting poses can be used for MD simulations59,80.
We also ran another set of simulations to corroborate the blind docking with other molecular docking software, including AutoDock Vina63, rDock66 and FlexAID67 (Figure 7). All three programs, which each use different algorithms, suggest that there are two potential binding sites. One is within the LBD, which corresponds to experimental data18, and the other is within the ‘bridge’. PCA of the results from these docking programs was performed for easier comprehension (Figure S12, Extended data26).
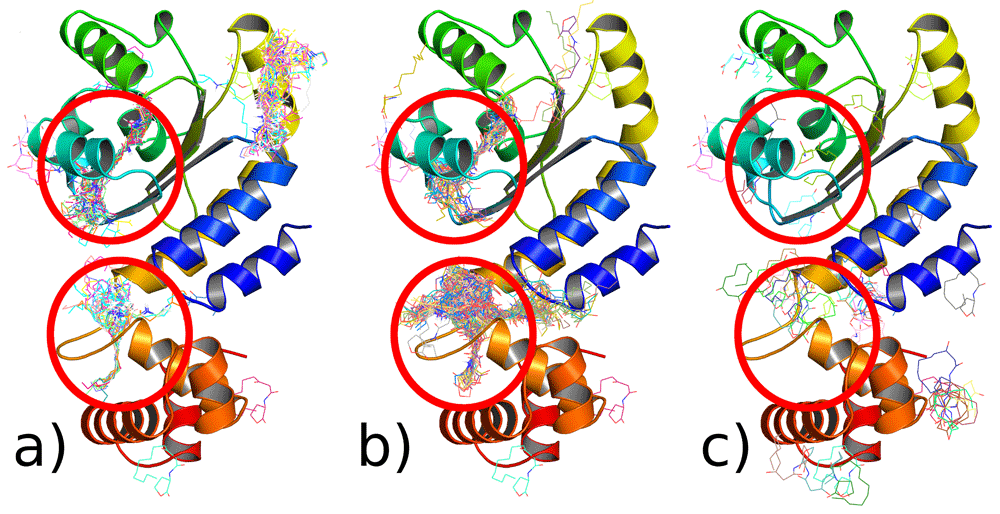
Red circles - binding interactions that are characteristic to all programs a) Autodock Vina b) rDock c) FlexAid.
In total, 900 nanoseconds of MD simulations have been performed and used for the analysis of 3OC12-HSL interaction with the LasR monomer. The representative structures were taken from docking results (Figure 6c) and used as starting points for MD simulations with LasR. Simulations were conducted for a sufficient time to allow the positions of 3OC12-HSL to reach equilibrium in the LasR molecule.
The stability of the molecule was evaluated by calculating the RMSD of the backbone atoms. RMSD was calculated with reference to the initial snapshot. Figure 8a shows that Simulation 2 and 3 experience a substantial RMSD deviation from the initial starting point. Simulation 2 corresponds to cluster 2 in the docking simulations, while Simulation 3 corresponds to cluster 3 (Figure 8a). For Simulation 1, which corresponds to cluster 1, the 3OC12-HSL molecule did not fixate and reach equilibrium and therefore no further analysis was performed (Figure 8a) for this simulation. Simulation 2 shows that after 230 nanoseconds, the structure becomes stable. For Simulation 3, the structure changes its conformation at 100 nanoseconds and becomes stable between 260 nanoseconds and 300 nanoseconds. Simulation 2 and 3 represent the binding sites predicted by all docking programs from Figure 7. Calculation of the root-mean-square fluctuation (RMSF) was used to evaluate the flexibility of the LasR monomer. The average per-residue RMSF was calculated for each simulation run (Figure 8b).
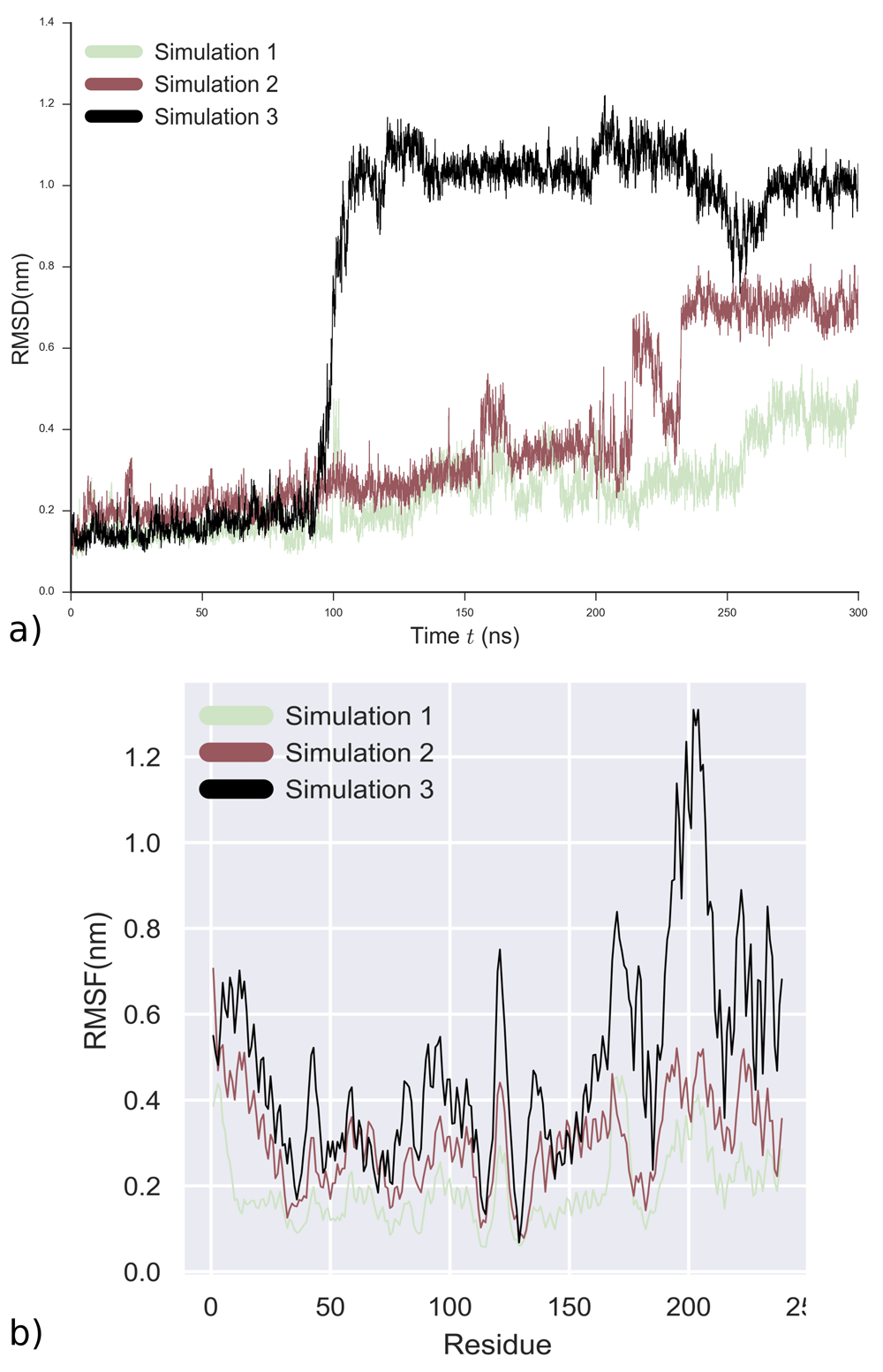
Analysis for 3 independent runs of 300 ns using representative structures from 3OC12-HSL docking poses as starting points a) RMSD evolution b) RMSF of Cα-atoms of LasR for each individual run.
It is visible in Figure 8b that the residues from 165 to 176, which correspond to beta turns in the SLR of LasR, are highly mobile. Simulations 2 and 3 show that 3OC12-HSL has two binding modes, one with the LBD (Figure 9a), which corresponds to experimental data and one with the LBD-SLR-DBD bridge of LasR (Figure 9d).
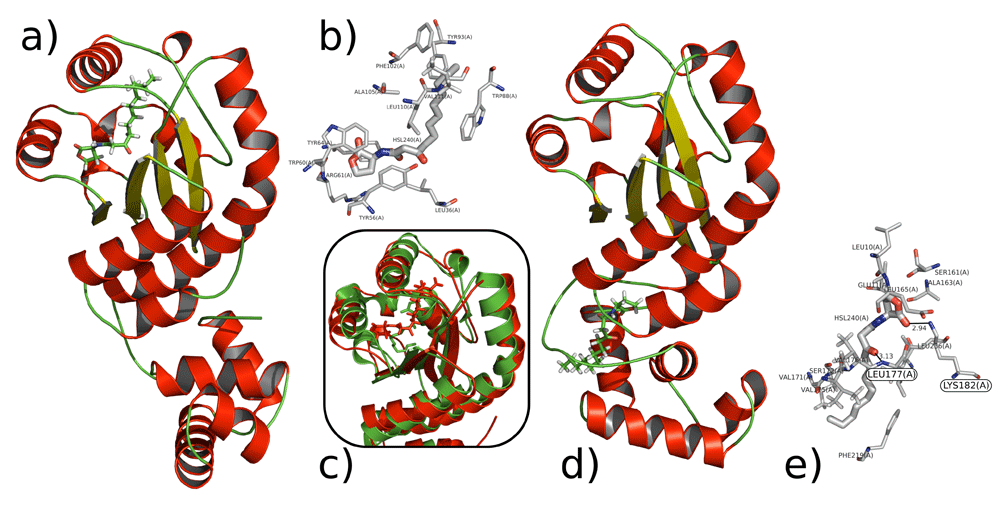
Visualizations of the interactions of 3OC12-HSL with LasR. Conformations taken from centroids from cluster analysis a) 3OC12-HSL with LasR LBD b) Insertion of 3OC12-HSL in LBD of LasR c) Superimposition of the modelled interaction (red) of 3OC12-HSL with LasR LBD and crystallographic structure (green) d) 3OC12-HSL with the “bridge” of LasR e) Putative hydrogen bonds of 3OC12-HSL with Lys182 and Leu177.
PCA and cluster analysis were performed on simulation 2. Hydrogen bond analysis was also performed, based on cut-offs for distance and angles according to the Wernet-Nilson criteria81 using MDTraj31. During the simulation, 3OC12-HSL forms hydrogen bonds with Tyr56, Ser129 and Trp60, which is in agreement with experimental data18 (Figure S14, Extended data26). Over the course of simulation 2, 3OC12-HSL also establishes a large number of hydrophobic contacts with the amino acid side chains of the LasR LBD (Figure 9b). This phenomenon is not unexpected, given the large hydrophobic surface area of the LasR LBD and the low solubility of 3OC12-HSL in water. 3OC12-HSL has hydrophobic interactions mainly with amino acids from helix 3, helix 5 and sheet 1 side chains. RMSD analysis of the conformations of LasR with and without 3OC12-HSL bound to the LBD gives a value of 7.027 Å between the conformations (Figure S16, Extended data26). RMSD of the modelled full structure of LasR with ligand superimposed onto the experimentally obtained tertiary structure of LBD of LasR with ligand gives a value of 1.692 Å, where RMSD <2 Å in relation to the experimentally studied structure (Figure 9c). Residues that participate in hydrophobic interactions and hydrogen bonding are shown in Figure 9e and Figure S18 and 19 (Extended data26).
The second binding mode involves the interaction of 3OC12-HSL with the LBD-SLR-DBD bridge. PCA, cluster and hydrogen bond analysis were also performed on simulation 3. Over the course of simulation 3, 3OC12-HSL establishes hydrogen bonds and a large number of hydrophobic contacts with amino acid side chains in the beta turns of the LasR SLR. In simulation 3, 3OC12-HSL forms hydrogen bonds mainly with the ‘Lys182’ and ‘Leu177’ beta turn residues. RMSD analysis of the two conformations of the LasR monomer and when LasR is bound to 3OC12-HSL via the DBD gives a value of 1.677 Å (Figure S20, see Extended data26). Residues that participate in hydrogen bonding and hydrophobic interactions are shown in Figure S16. Complete data sets for MD simulations are available as extended data (Figures S13-S2026).
In order to analyse the binding sites in detail, MM-PBSA69 binding energy calculations were performed for each binding site based on the MD trajectories. The binding energy calculations demonstrated that the interaction of the ligand with the LBD-SLR-DBD bridge is not competitive with its interaction with the LBD (Table 3). Analysis of the energy terms demonstrated that only polar solvation energy contributes positively. However, for the interaction with the “the bridge”, the electrostatic interaction energy is 3.6 times higher than the energy for the LBD interaction.
This suggests that a combination of Van der Waals, electrostatic interaction and non-polar solvation energy contribute to the stability of the LasR-3OC12-HSL binding complex. To assess which amino acids are involved, we performed analysis of the energy contribution of residues to binding for both simulations obtained from MM-PBSA calculations using g_mmpbsa. Sequence alignment was performed using the R msa package70. ClustalW71, Clustal Omega72 and Muscle73 algorithms were used for sequence alignment. Full alignments using these algorithms are available as extended data26. Analysis suggested that LasR residues Tyr64, Tyr56, Trp88, Leu36 and Trp60 contribute the most to binding with 3OC12-HSL (Figure 10a), with a binding energy of approximately -128.578 kJ/mol (Table 3).
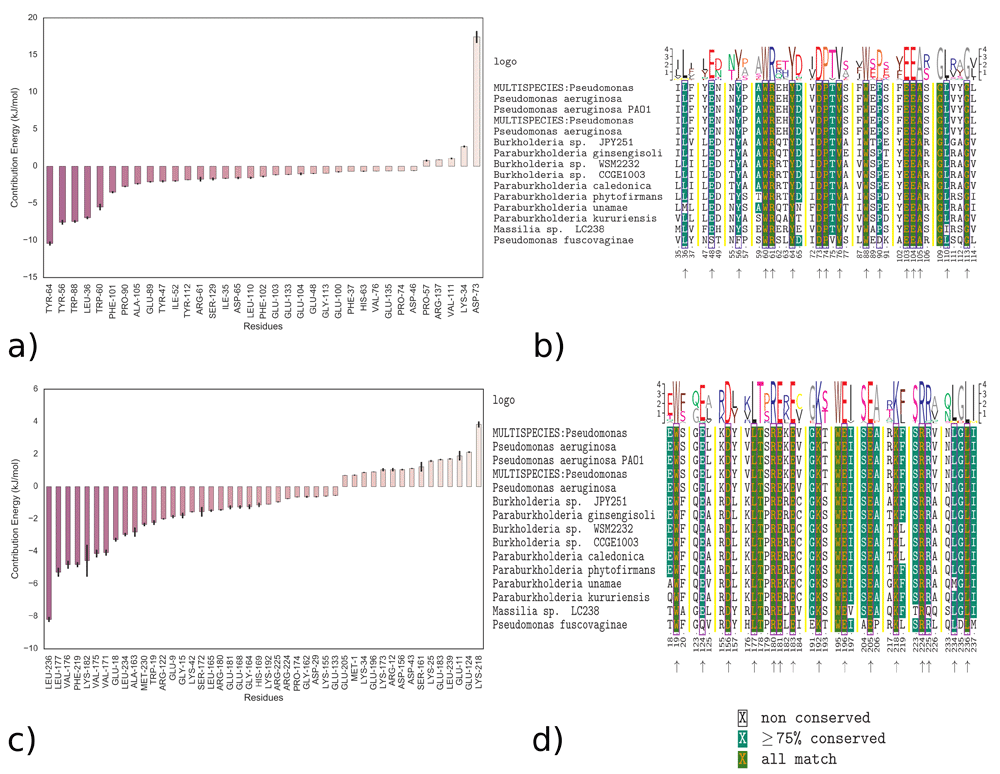
Energy contributions of residues and multiple sequence alignments demonstrating the conserved amino acid residues of LasR that interact with 3OC12-HSL: a) All amino acids that interact with LBD b) Conserved amino acids that interact with LBD. Blue boxes and arrows point the amino acid residues that interact with 3OC12-HSL c) All amino acids that interact with the “bridge” d) Conserved amino acids that interact with “bridge”. Black boxes and arrows point the residues that interact with 3OC12-HSL.
Energy calculations to assess residue interactions and sequence alignment show that 3OC12-HSL interacts with highly conserved amino acid Trp60, which agrees with experimental data82. The interaction also involves nine other conserved amino acids; Tyr64, Asp73, Pro74, Val76, Phe102, Phe103, Ala105 and Gly113 (Figure 10b). In total, 16 amino acids that contribute to the interaction have over 75% conservation.
Analysis suggested that ligand residues Leu236, Leu177, Val176, Phe219, Lys182 and Trp19 contribute the most (Figure 10c) to the newly described binding state involving ‘the bridge’ of LasR, with a binding energy of approximately -166.456 kJ/mol (Table 3). The analysis of residue sequence alignment shows that 3OC12-HSL interacts with highly conserved amino acids such as Leu236, Leu177, Phe219 and Trp19 (Figure 10d). The 3OC12-HSL interacts with 12 fully conserved amino acids in this case. In total, 16 amino acids participate in the interaction, where amino acid conservation is more than 75%. In this new binding site, Trp19 and Asp156 of the LBD, Leu177 of the SLR and Arg180, Glu181, Glu196, Lys218, Arg224 and Lys236 of the DBD of LasR participate in the interactions with 3OC12-HSL. A lactone head group interacts with the conserved Trp19 of the LBD.
It is known that LasR binds to the corresponding promoter as a dimer1 after interacting with the autoinducer. Thus, monomeric LasR–3OC12-HSL complexes were subjected to dimerization as an additional experiment. The protein docking experiments using structures from MD runs were performed using ClusPro v2.074,83–85 because of its success in the CAPRI (Critical Assessment of Predicted Interactions) scoring experiment. We used the centroid conformations of the LasR bridge-3OC12-HSL and LasR LBD-3OC12-HSL complexes from MD simulations. Then we performed protein docking using protein conformations without the ligands and used constraints based on the residue distances between monomers from the experimental data18. For the selection of the model, we used an approach recommended by the authors of ClusPro74, which suggests finding the most populated clusters. For LasR bridge-3OC12-HSL complex docking, the top model contained 80 members and the scores by ClusPro for the docking model were -951.4 for the centre and -1332.0 and for the lowest energy region, suggesting a favourable binding mode (Figure 11a). For the LBD LasR-3OC12-HSL complex, the top model contained 122 members and the scores for the docking model were -1440.7 for the centre and -1517.9 for the lowest energy region (Figure 11b). Both docking structures feature negative energy and this could suggest the possibility of multiple docking conformations. It is possible that the multiple binding modes of 30C12-HSL imply that LasR has multiple dimerization interfaces like QscR (Figure 11c)20, which is a post-translational repressor of LasR. In the absence of 3OC12-HSL, it forms a heterodimer with LasR. It appears this protein may have a much more delicate regulation than was initially thought.
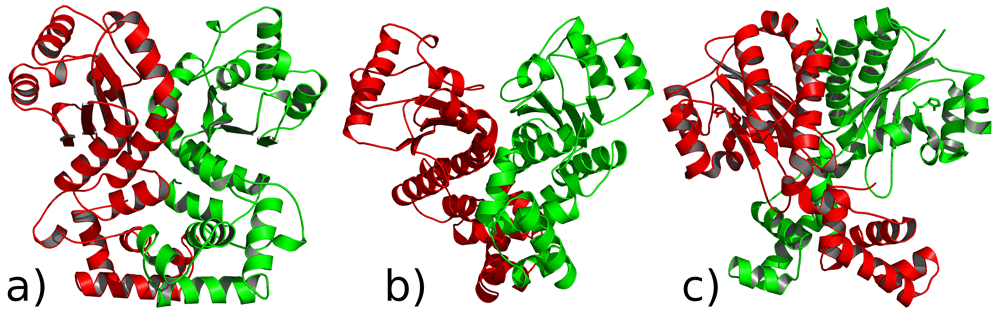
LasR monomer protein docking results and crystallographic dimer structure of quorum sensing control repressor QscR bound to 3OC12-HSL a) using centroid conformation from the LBD LasR-3OC12-HSL complex b) using centroid conformation from the “bridge”-3OC12-HSL complex c) crystallographic dimer structure of QscR, which is a homologue of LasR.
Pseudomonosa aeruginosa is one of the ESKAPE pathogens3, which are the leading cause of nosocomial infections throughout the world. However, the molecular mechanisms of the LasR protein in regulating quorum sensing machinery have not been fully elucidated. LasR has been problematic to structurally analyse for the last decade because of the insolubility of the full protein18. All of the available crystallographic structures contain only the LBD and lack the DBD. The lack of a full crystal structure has prevented us from furthering our understanding of LasR as knowledge of its full 3D structure is vital. It is therefore very important to study the interaction of the autoinducer with the full structure of the LasR protein. Adequate knowledge of this interaction would provide the opportunity to conduct improved structure-based drug discovery studies.
Results from our analysis show that the interaction of 3OC12-HSL with LasR has two binding modes. The results were cross-checked using multiple docking programs, molecular dynamics simulations and machine learning techniques. The 3OC12-HSL has a long hydrophobic tail and is therefore capable of binding to both the LBD and the “bridge” of LasR. These results suggest that both the C-terminal and the N- terminal regions of LasR interact with 3OC12-HSL. The binding of 3OC12-HSL provokes conformational transitions in the structure of LasR. Binding energy analysis showed that the “bridge” does not compete with LBD. An MM-PBSA approach was used for the calculation of relative binding energy69, although this finding needs confirmation through further research. The calculation of absolute binding energy could give more reliable results, though this type of calculation is more time consuming62. The results of our in silico experiments for the interaction of 3OC12-HSL with the LasR LBD is comparable with the results of other authors18,82, although currently there are no studies investigating the DBD.
This work is connected to recent experimental studies concerning LasR regulation. It should be noted that the LasR quorum-sensing system of P. aeruginosa is regulated by pre-quorum and post-quorum regulating systems86. The first system is controlled by post-translational repressors QscR, QslA and QteE20,86–89. In the absence of 3OC12-HSL, they can each form a heterodimer with LasR. Structural and functional studies of bacterial QS anti-activators revealed three different modes of action for LuxR-type transcriptional regulation: the destruction of the LBD dimerization interface in P. aeruginosa, the occupation of the AHL binding pocket and binding to the DBD11. All these interactions indirectly change the conformation of the DBD, thus allosterically preventing DNA binding. It was previously shown that QslA can bind to LasR and thereby inhibit its DNA-binding capability86. Moreover, it has been shown that QslA supresses formation of the LasR homodimer complex by occupying the same surface area that is used for dimerization88,89.
It has also been shown that QscR interacts with 3OC12-HSL20. QscR has multiple dimerization interfaces and possesses both an LBD and DBD20. The heterodimer QslA-LasR affects the dimerization interface and prevents receptor activation and DNA promoter binding88. According to one of the mechanisms, the anti-activator QslA blocks the regions that are necessary for transcription factors multimerization15. Moreover, 3OC12-HSL does not overcome the repression of LasR exerted by QslA. This indicates that 3OC12-HSL interacts with helixes that form part of these dimerization interfaces. It may be that the multiple binding modes of 30C12-HSL imply that LasR has multiple dimerization interfaces like QslA and QscR.
There is also another aspect to be discussed. One major strategy for LasR inhibition is the development of small-molecule antagonists that mimic the native autoinducer and inactivate LasR. However, the mechanism by which they inactivate LasR is unknown90. It has been suggested that LasR interactions with these antagonists promote a robust, unnatural fold of LasR that does not permit DNA binding90. Further research studying flavonoids as potential inhibitors shows that they do not bind in the ligand-binding pocket of LasR91. It has also been shown that they prevent LasR from binding to promoter DNA, but do not stop dimerization91. Unfortunately, in this study it was not possible to assay the LasR DBD due to insufficient protein yields91. The author suggest that the AI and flavonoid inhibitors can simultaneously bind to LasR and therefore postulate that flavonoids do not use the canonical AI binding site. We suggest that flavonoids and other inhibitors may interact with the bridge, thereby preventing LasR from binding to promoter DNA.
Our model might explain the molecular mechanisms proposed by recent experimental studies involving the study of flavonoids and other inhibitors which suggest the possibility of another binding site90,91. It may also explain why computational drug design has not been successful in targeting this protein. However, the lack of a full, experimentally-derived structure for LasR is problematic and requires further experimental and computational research. We expect that our molecular insights on LasR can shed more light on this protein and assist in the development of new treatments against P. aeruginosa.
From the simulations, it appears that the AI ligand 3OC12-HSL can bind to both the LBD and the “bridge” of the transcriptional regulator LasR. The interaction with the “bridge” is a novel site. Binding energy analysis shows that the interaction of 3OC12-HSL with the “bridge” and its interaction with the LBD is not competitive. Conserved amino acids such as Leu236, Phe219, Leu177, Lys182 and Trp19 contribute the most during the interaction with “bridge”. This could suggest that the interaction of 3OC12-HSL with the “bridge” is necessary for the DNA binding capability of LasR. One possible explanation for these multiple binding sites is that LasR may have multiple dimerization interfaces. This study may reveal new insights into the interactions of the native 3OC12-HSL ligand with transcriptional regulator LasR in P. aeruginosa. Results from this study may aid future drug development endeavours.
All data underlying the results are available as part of the article and no additional source data are required.
Zenodo: Interaction of N-3-oxododecanoyl homoserine lactone with transcriptional regulator LasR of Pseudomonas aeruginosa: Insights from molecular docking and dynamics simulations. https://doi.org/10.5281/zenodo.258655926
This project contains the following extended data:
- AdditionalFile1_SupportingInformation.pdf (Figures S1-20)
- AdditionalFile2_ClustalW.pdf (Full sequence alignment of LasR using ClustalW algorithm)
- AdditionalFile3_ClustalOmega.pdf (Full sequence alignment of LasR using Clustal Omega algorithm)
- AdditionalFile4_Muscle.pdf (Full sequence alignment of LasR using Muscle algorithm)
- HSL_dock.zip (Molecular docking simulations input and output for AutoDock Vina)
- HSL_exhaustiveness.zip (Molecular docking simulations input and output for the determination of exhaustiveness (AutoDock Vina))
- LasR_HSL_multidock.zip (Molecular docking simulations input and output for AutoDock Vina, rDock and FlexAID)
- MD100ns.tar.gz (Molecular dynamics simulation data of LasR protein for 100 nanoseconds)
- HSL_LasR_simulation_1.zip (1st MD simulations data for 3OC12-HSL with LasR) HSL_LasR_simulation_2.zip (2nd MD simulation data for 3OC12-HSL with LasR)
- HSL_LasR_simulation_3.zip (3rd MD simulation data for 3OC12-HSL with LasR)
- scripts.zip (Scripts that were used for the analysis of the data based on the molmolpy set of scripts92)
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)