1Department of Dermatology, Yale University School of Medicine, New Haven, CT, USA 2Department of Pathology, Yale University School of Medicine, New Haven, CT, USA 3Department of Genetics, Yale University School of Medicine, New Haven, CT, USA
Vascular tumors are neoplasms of endothelial cells, a significant number of which present in childhood. Recent studies have examined the mutational landscape of many subtypes of vascular tumors, identifying mutations primarily within the Ras–mitogen-activated protein kinase (MAPK) pathway and providing a unique opportunity to consider targeted therapeutics. This review will summarize the current understanding of childhood vascular tumor pathobiology.
Corresponding author:
Keith Choate
Competing interests:
No competing interests were disclosed.
Grant information:
The work for this report was supported by the Leon Rosenberg, M.D., Medical Student Research Fund in Genetics and the Jane Danowski Weiss Family Foundation Fellowship at the Yale University School of Medicine.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Vascular anomalies are divided into two groups: malformations and tumors1. The most recent International Society for the Study of Vascular Anomalies (ISSVA) classification for vascular tumors further subdivides them into three primary groups: benign tumors, locally aggressive or borderline tumors, and malignant tumors (Table 1).
Table 1. International Society for the Study of Vascular Anomalies (ISSVA) classification of vascular tumors23.
Infantile hemangioma (IH) is the most common childhood vascular neoplasm; IH has an incidence of approximately 4.5% by 3 months of age2. However, the genetic mechanism of IH pathobiology remains unknown despite its prevalence. In 1999, Walter et al. mapped a familial form of IH to 5q31-33, housing three candidate genes—fibroblast growth factor receptor-4 (FGFR4), platelet-derived growth factor receptor-β (PDGF-β), and fms-related tyrosine kinase-4 (FLT4)3—and in subsequent work found that a small number of IHs (2 out of 15 studied cases) harbor somatic mutations in vascular endothelial growth factor (VEGF)-receptor 2 (p.P1147S) and VEGFR3 (p.P954S) (also known as FLT4)4. Nonetheless, these variants have yet to be confirmed via in vitro or in vivo studies to cause vascular tumors or oncogenic transformation. However, a recent single-nucleotide polymorphism (SNP) study of VEGFR-2 and VEGF-A in IH was unable to detect variants associated with disease, although the G allele of rs2010963 in VEGF-A was associated with a significantly lower risk of IH5. Additionally, there is some debate regarding the sporadic or familial etiology of IH. Whereas twin studies suggest extra-genetic factors as the primary cause of disease, recent work studying multiple pedigrees suggests an either autosomal dominant or maternally transmitted inheritance pattern6,7. Genetic analysis of syndromic forms of IH, including PHACE syndrome (posterior fossa malformations, infantile hemangiomas, arterial anomalies, cardiac defects, and eye anomalies syndrome), which appears more often in female offspring, suggests a possible X-linked recessive pattern but has not identified a somatic mutation associated with most cases8–10. Similarly, a causative mutation has not yet been identified in LUMBAR syndrome (lower body hemangioma, urogenital malformation, myelopathy, bony deformities, anorectal malformations, arterial anomalies, and renal anomalies syndrome11. The three leading hypotheses on the pathogenesis of IH are (1) local hypoxemia leading to hypoxia-inducible factor 1 alpha (HIF-1α)-induced proliferation12,13, (2) embolization of placental cells14–17, and (3) vasculogenesis/angiogenesis driven by hypoxemia-induced differentiation of mesenchymal stem cells into endothelial cells and Notch-mediated differentiation of mesenchymal stem cells into proangiogenic pericytes18–22. These hypotheses do not address the multi-system defects found in PHACE or LUMBAR syndromes which suggest that somatic mosaicism plays a role in pathogenesis.
Unlike other childhood vascular tumors, IH responds to beta-blockers; 60% of patients experience complete or near-complete resolution of the lesion and 88% of patients demonstrate improvement following a 6-month course of propanolol at a dose of 3 mg/kg per day24. Prior to the advent of beta-blockers in the treatment of IH, treatment with systemic corticosteroids was considered the standard of care, and a pooled meta-analysis estimated that 69% of lesions respond to therapy25, although significant morbidity—including Cushingoid features, gastroesophageal reflux, hypertension, ulceration, bleeding, failure to thrive, hirsutism, hypercholesterolemia, and infection—was also reported26,27. IH can also be distinguished from other tumors by its positive GLUT1 immunoreactivity; up to 97% of lesions show positive signal17,28. Notably, however, about half of the vessels in a given tumor are GLUT1-negative, suggesting that a heterogeneous population of endothelial cells populates these lesions, an idea later confirmed by in vitro studies of cells isolated from IH samples17,29. Although the genetic factors contributing to IH pathobiology remain unknown, many somatic mutations associated with GLUT1-negative vascular tumors have been identified in recent years, most in genes already known to be implicated in tumorigenesis.
IDH1/IDH2
Genetic insight into spindle cell hemangiomas (SCHs) came from studies of Maffucci syndrome (Spranger type II enchondromatosis), a subtype of enchondromatosis presenting with multiple SCHs in early childhood30. In an analysis of 13 patients with Maffucci syndrome, Pansuriya et al. found that 70% of SCHs had p.R132C mutations in exon 4 of isocitrate dehydrogenase 1 (IDH1)31. Given the disorder’s unilateral distribution of the endochondromas, the lack of mutations detected in adjacent non-lesional tissue, the absence of transmission within pedigrees, and the identification of tissue-specific IDH1 mutations, somatic mosaicism was considered causal. Further work in sporadic, acquired SCH found that IDH1 p.R132C is found in at least 64% of cases32. Among cases negative for IDH1 p.R132C, 20% had mutations at arginine 172 in exon 4 of IDH2, suggesting genetic heterogeneity32.
Mutations in exon 4 at arginine 132 of IDH1 or at arginine 140 or 172 of IDH2 lead to the production of 2-hydroxyglutarate, an oncometabolite which causes a hypermethylation phenotype leading to the inhibition of genes responsible for terminal differentiation33–36. Additionally, mutations in IDH1 found in gliomas lead to reduction of alpha-ketoglutarate production, inducing HIF-1α, which drives tumor growth via the hypoxia pathway37. Interestingly, analysis of HIF-1α in SCH revealed a lack of expression in all samples32, suggesting that IDH1 and IDH2 mutations driving SCH may act via a distinct mechanism.
CAMTA1/TFE
Of childhood vascular tumors, epithelioid hemangioendothelioma (EHE) is the most common malignant variety. In 2001, Mendlick et al. reported an identical chromosomal translocation of t(1;3) (p36.3;q25) in two cases of EHE38. Owing to low tumor cellularity and the absence of EHE cell lines, the specific genes disrupted via the translocation remained unknown until 2011, when Tanas et al. employed RNA sequencing to identify a fusion between the promotor region of WW domain-containing transcription regulator 1 (WWTR1) on 3q25 and the carboxyl terminus of calmodulin-binding transcription activator 1 (CAMTA1) on 1p3639. Given the high activity of the WWTR1 promoter in endothelial cells and the ectopic expression of CAMTA1, which is typically found only in brain tissue, the authors hypothesized that WWTR1/CAMTA1 functions as an oncogene via a promoter switch mechanism. Further work found that the WWTR1-CAMTA1 fusion is a consistent genetic finding in EHEs of different anatomic subsites40.
In EHE samples without a WWTR1-CAMTA1 mutation, a distinct gene fusion between transcription factor E3 (TFE3) and yes-associated protein 1 (YAP1) was identified41. Given the structural and functional similarities between YAP1 and WWTR1 as well as the oncogenic nature of TFE3 with preserved transcriptional activation domains, well recognized in other cancers42–44, a promoter switch similar to that of WWTR1-CAMTA1 fusions is hypothesized to underlie oncogenesis in cases with YAP1-TFE3 fusions.
GNA family
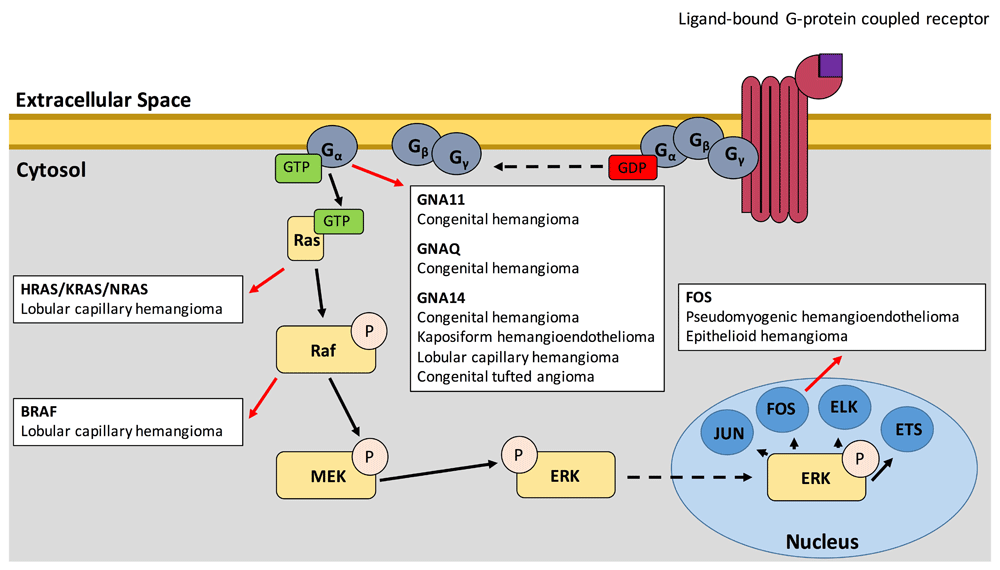
In recent years, a number of studies have highlighted the importance of the Ras–mitogen-activated protein kinase (MAPK) pathway in the oncogenic transformation of many childhood vascular tumors (Figure 1). The most upstream portion of the pathway elucidated thus far is the guanine nucleotide-binding protein subunit alpha q (Gαq) family of genes: GNAQ, GNA11, and GNA14. In 2016, three studies demonstrated that somatic activating mutations in these genes are found in congenital hemangioma (including both rapidly involuting congenital hemangiomas and non-involuting congenital hemangiomas), kaposiform hemangioendotheliomas (KHEs), congenital tufted angiomas (TAs), and childhood lobular capillary hemangiomas (LCHs) via whole-exome sequencing45–47. Activating mutations at the arginine 183 position of GNA11 and the glutamine 209 position of GNA11 and GNAQ were also found in several cases of sporadic congenital hemangioma, while mutation of glutamine 205 in GNA14, the analogous position of glutamine 209 in GNA11 and GNAQ, was found in one case each of KHE, LCH, and TA45,46. Recent work has also identified mutations in the glutamine 205 position of GNA14 and the glutamine 209 position of GNAQ in anastomosing hemangiomas48. Although these mutations have been demonstrated to cause cell morphology changes, upregulation of biochemical growth pathways, and acquisition of growth factor independence in vitro, they have not yet been shown to cause disease in an in vivo model. Notably, arginine 183 mutation in GNAQ is thought to be responsible for up to 88% of Sturge–Weber syndrome which presents with port-wine stains (PWSs) and leptomeningeal vascular malformations, while similar GNA11 and GNAQ activating mutations underlie 77% of capillary malformations, 46% of uveal melanomas, and 83% of blue nevi49–52. Activated GNA11 and GNAQ are thought to mediate VEGFR-2 phosphorylation, triggering human umbilical vein endothelial cell (HUVEC) proliferation in vitro53.
Figure 1. Positions of the Ras-MAPK pathway wherein mutations have been demonstrated to lead to childhood vascular tumors (red arrows).
Unbroken arrows indicate activation while broken arrows indicate migration.
These Gα-subunit proteins exchange bound GDP for GTP when their associated G protein–coupled receptor (GPCR) is activated54,55, leading to dissociation of the G-protein heterotrimer—composed of Gα, Gβ, and Gγ—from the GPCR and of the activated Gα subunit from the Gβ/Gγ dimer, ultimately leading to the downstream activation of several cellular pathways.
The mechanism of tumorigenesis associated with these mutations can provide insights into targeted therapeutics. Gα is upstream of both the Ras-MAPK and the PI3K-Akt-mTOR, and previous studies of low-flow, lymphatic/venous, vascular malformations implicated the PI3K-Akt-mTOR pathway as the primary driver of these lesions56–59. Although sirolimus is effective in the treatment of these low-flow lesions60–63, it has shown little efficacy in vascular tumors, suggesting a distinct pathobiology62,64,65. In a study using primary HUVECs expressing mutant GNA11 and GNA14, cells showed no indication of PI3K-Akt-mTOR pathway activation (as measured by pAKT) and instead showed specific activation of the Ras-MAPK pathway (via increased pERK)47, suggesting that a more effective therapy may involve targeting the Ras-MAPK pathway. Recent work suggests that high-flow arteriovenous malformations (AVMs) are also driven by mutations within the Ras-MAPK signaling pathway66. Thus, advances in therapy for vascular tumors may also benefit patients with these high-flow vascular malformations. Somatic mutations in downstream components of this pathway in other vascular tumors further highlight the Ras-MAPK pathway as a primary driver of tumorigenesis in childhood vascular tumors.
The MAPK pathway
Activation of Gα leads to increased RAS activation67. Indeed, a study of sporadic LCHs identified somatic mutations in all three subgroups of the RAS subfamily: HRAS, KRAS, and NRAS68. The activating mutations, which mainly fall at codons 12, 13, and 61, have been established to generate constitutive Ras-MAPK signaling by preventing GTP hydrolysis69. Furthermore, a study of LCHs arising within PWS also identified a p.V600E mutation in BRAF, a proto-oncogene directly downstream of Ras in the MAPK pathway chain70. Interestingly, the study also found that both the underlying PWS and the LCH carried mutations in the arginine 183 position of GNAQ but that BRAF or RAS mutation was specific to the LCHs, suggesting that the LCHs arose because of a “second-hit” in RAS or BRAF.
Mutations in the Fos family of transcription factors, which are among the final components of the Ras-MAPK pathway, have also been identified in childhood vascular tumors. Cytogenetic study of pseudomyogenic hemangioendothelioma (PHE) revealed a t(7;19) (q22;q13) translocation as the sole anomaly in three lesions from one patient71. Later study of this patient and an additional case of PHE revealed that this translocation leads to a SERPINE1-FOSB fusion gene72. Although vascular endothelial cells demonstrate strong endogenous expression of SERPINE1, the SERPINE1-FOSB disrupts the protein-coding portion of the SERPINE1 gene, instead generating high levels of FOSB mRNA via a promoter switch mechanism72–74. FOSB and FOS mutations have also been identified in cases of epithelioid hemangioma (EH). In one study of EH, Antonescu et al. identified two fusion genes: WWTR1/FOSB and ZFP36/FOSB75. It has also been demonstrated that the FOS rearrangement leading to the truncation of the FOS protein, specifically to loss of the transactivation domain (TAD), leads to EH in bone.
Summary
The highlighted genetic discoveries in vascular tumor biology provide novel targets for therapeutics. Indeed, the fact that most of these mutations are present in known cancer-causing pathways means that many medications that are currently approved or under trial for other malignancies may be repurposed for use in vascular tumor therapy.
Mutations in IDH are known to cause gliomas, glioblastomas, chondrosarcomas, intrahepatic cholangiocarcinomas, and hematologic malignancies in addition to SCHs76. As such, a number of therapeutics currently under investigation may also be effective in the treatment of SCH. Early results from trials of ivosidenib (AG-120), a novel inhibitor of mutant IDH1, in IDH1-mutated acute myelogenous leukemia (AML) indicated an overall response rate of 41.6% and a complete remission rate of 21.6%77. Similarly, results from early trials of enasidenib (AG-221), a novel inhibitor of mutant IDH2, in IDH2-mutated AML indicated an overall response rate of 40.3% and a complete remission rate of 19.3%78. These agents are currently under study in a number of clinical trials (ClinicalTrials.gov Identifiers: NCT02074839, NCT02073994, NCT01915498, NCT02577406, NCT02632708, and NCT02677922). Mouse studies have also shown potential for an IDH1 peptide vaccine79, which is currently under trial (ClinicalTrials.gov Identifiers: NCT02454634 and NCT02193347).
Selective inhibition of the Ras-MAPK pathway may provide a novel therapeutic avenue for childhood vascular lesions, which currently have few effective non-surgical options80. The central role of this pathway in tumor pathobiology has necessitated the development of a number of currently available medications, including farnesyl transferase inhibitors such as salirasib, BRAF inhibitors such as vemurafenib, MEK inhibitors such as trametinib, and ERK inhibitors such as ulixertinib, which warrant further study as therapy for childhood vascular tumors. Indeed, Al-Olabi et al. demonstrated that treatment of AVMs in BRAF-mutant zebrafish with vemurafenib leads to restoration of blood flow in AVMs where it was previously limited81. With promising early results, these therapies hold great potential for the treatment of childhood vascular tumors.
Grant information
The work for this report was supported by the Leon Rosenberg, M.D., Medical Student Research Fund in Genetics and the Jane Danowski Weiss Family Foundation Fellowship at the Yale University School of Medicine.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Faculty Opinions recommended
References
1.
Mulliken JB, Glowacki J:
Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics.
Plast Reconstr Surg.
1982; 69(3): 412–22. PubMed Abstract
| Publisher Full Text
2.
Kanada KN, Merin MR, Munden A, et al.:
A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature.
J Pediatr.
2012; 161(2): 240–5. PubMed Abstract
| Publisher Full Text
3.
Walter JW, Blei F, Anderson JL, et al.:
Genetic mapping of a novel familial form of infantile hemangioma.
Am J Med Genet.
1999; 82(1): 77–83. PubMed Abstract
| Publisher Full Text
4.
Walter JW, North PE, Waner M, et al.:
Somatic mutation of vascular endothelial growth factor receptors in juvenile hemangioma.
Genes Chromosomes Cancer.
2002; 33(3): 295–303. PubMed Abstract
| Publisher Full Text
8.
Frieden IJ, Reese V, Cohen D:
PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities.
Arch Dermatol.
1996; 132(3): 307–11. PubMed Abstract
| Publisher Full Text
9.
Siegel DH, Shieh JTC, Kwon EK, et al.:
Copy number variation analysis in 98 individuals with PHACE syndrome.
J Invest Dermatol.
2013; 133(3): 677–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
11.
Iacobas I, Burrows PE, Frieden IJ, et al.:
LUMBAR: association between cutaneous infantile hemangiomas of the lower body and regional congenital anomalies.
J Pediatr.
2010; 157(5): 795–801.e7. PubMed Abstract
| Publisher Full Text
12.
Kleinman ME, Greives MR, Churgin SS, et al.:
Hypoxia-induced mediators of stem/progenitor cell trafficking are increased in children with hemangioma.
Arterioscler Thromb Vasc Biol.
2007; 27(12): 2664–70. PubMed Abstract
| Publisher Full Text
13.
Chang EI, Chang EI, Thangarajah H, et al.:
Hypoxia, Hormones, and Endothelial Progenitor Cells in Hemangioma.
Lymphat Res Biol.
2007; 5(4): 237–43. PubMed Abstract
| Publisher Full Text
14.
Barnés CM, Huang S, Kaipainen A, et al.:
Evidence by molecular profiling for a placental origin of infantile hemangioma.
Proc Natl Acad Sci U S A.
2005; 102(52): 19097–102. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
Barnés CM, Christison-Lagay EA, Folkman J:
The placenta theory and the origin of infantile hemangioma.
Lymphat Res Biol.
2007; 5(4): 245–55. PubMed Abstract
| Publisher Full Text
16.
Mihm MC Jr, Nelson JS:
Hypothesis: The metastatic niche theory can elucidate infantile hemangioma development.
J Cutan Pathol.
2010; 37 Suppl 1: 83–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
17.
North PE, Waner M, Mizeracki A, et al.:
GLUT1: A newly discovered immunohistochemical marker for juvenile hemangiomas.
Hum Pathol.
2000; 31(1): 11–22. PubMed Abstract
| Publisher Full Text
20.
Boscolo E, Stewart CL, Greenberger S, et al.:
JAGGED1 Signaling Regulates Hemangioma Stem Cell–to–Pericyte/Vascular Smooth Muscle Cell Differentiation.
Arterioscler Thromb Vasc Biol.
2011; 31(10): 2181–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
21.
Wu JK, Adepoju O, De Silva D, et al.:
A switch in Notch gene expression parallels stem cell to endothelial transition in infantile hemangioma.
Angiogenesis.
2010; 13(1): 15–23. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Boscolo E, Mulliken JB, Bischoff J:
Pericytes from infantile hemangioma display proangiogenic properties and dysregulated angiopoietin-1.
Arterioscler Thromb Vasc Biol.
2013; 33(3): 501–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
23.
Wassef M, Blei F, Adams D, et al.:
Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies.
Pediatrics.
2015; 136(1): e203–14. PubMed Abstract
| Publisher Full Text
24.
Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al.:
A randomized, controlled trial of oral propranolol in infantile hemangioma.
N Engl J Med.
2015; 372(8): 735–46. PubMed Abstract
| Publisher Full Text
25.
Izadpanah A, Izadpanah A, Kanevsky J, et al.:
Propranolol versus corticosteroids in the treatment of infantile hemangioma: a systematic review and meta-analysis.
Plast Reconstr Surg.
2013; 131(3): 601–13. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
26.
Price CJ, Lattouf C, Baum B, et al.:
Propranolol vs corticosteroids for infantile hemangiomas: a multicenter retrospective analysis.
Arch Dermatol.
2011; 147(12): 1371–6. PubMed Abstract
| Publisher Full Text
27.
Pandey A, Gangopadhyay AN, Gopal SC, et al.:
Twenty years' experience of steroids in infantile hemangioma--a developing country's perspective.
J Pediatr Surg.
2009; 44(4): 688–94. PubMed Abstract
| Publisher Full Text
28.
North PE, Waner M, Mizeracki A, et al.:
A unique microvascular phenotype shared by juvenile hemangiomas and human placenta.
Arch Dermatol.
2001; 137(5): 559–70. PubMed Abstract
29.
Huang L, Nakayama H, Klagsbrun M, et al.:
Glucose Transporter 1-Positive Endothelial Cells in Infantile Hemangioma Exhibit Features of Facultative Stem Cells.
Stem Cells.
2015; 33(1): 133–45. PubMed Abstract
| Publisher Full Text
| Free Full Text
30.
Pansuriya TC, Kroon HM, Bovée JV:
Enchondromatosis: insights on the different subtypes.
Int J Clin Exp Pathol.
2010; 3(6): 557–69. PubMed Abstract
| Free Full Text
31.
Pansuriya TC, van Eijk R, d'Adamo P, et al.:
Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome.
Nat Genet.
2011; 43(12): 1256–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
32.
Kurek KC, Pansuriya TC, van Ruler MA, et al.:
R132C IDH1 mutations are found in spindle cell hemangiomas and not in other vascular tumors or malformations.
Am J Pathol.
2013; 182(5): 1494–500. PubMed Abstract
| Publisher Full Text
38.
Mendlick MR, Nelson M, Pickering D, et al.:
Translocation t(1;3)(p36.3;q25) is a nonrandom aberration in epithelioid hemangioendothelioma.
Am J Surg Pathol.
2001; 25(5): 684–7. PubMed Abstract
| Publisher Full Text
39.
Tanas MR, Sboner A, Oliveira AM, et al.:
Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma.
Sci Transl Med.
2011; 3(98): 98ra82. PubMed Abstract
| Publisher Full Text
40.
Errani C, Zhang L, Sung YS, et al.:
A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites.
Genes Chromosomes Cancer.
2011; 50(8): 644–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
41.
Antonescu CR, Le Loarer F, Mosquera JM, et al.:
Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma.
Genes Chromosomes Cancer.
2013; 52(8): 775–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
42.
Sidhar SK, Clark J, Gill S, et al.:
The t(X;1)(p11.2;q21.2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene.
Hum Mol Genet.
1996; 5(9): 1333–8. PubMed Abstract
| Publisher Full Text
43.
Clark J, Lu YJ, Sidhar SK, et al.:
Fusion of splicing factor genes PSF and NonO (p54nrb) to the TFE3 gene in papillary renal cell carcinoma.
Oncogene.
1997; 15(18): 2233–9. PubMed Abstract
| Publisher Full Text
44.
Ladanyi M, Lui MY, Antonescu CR, et al.:
The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25.
Oncogene.
2001; 20(1): 48–57. PubMed Abstract
| Publisher Full Text
45.
Funk T, Lim Y, Kulungowski AM, et al.:
Symptomatic Congenital Hemangioma and Congenital Hemangiomatosis Associated With a Somatic Activating Mutation in GNA11.
JAMA Dermatol.
2016; 152(9): 1015–20. PubMed Abstract
| Publisher Full Text
47.
Lim YH, Bacchiocchi A, Qiu J, et al.:
GNA14 Somatic Mutation Causes Congenital and Sporadic Vascular Tumors by MAPK Activation.
Am J Hum Genet.
2016; 99(2): 443–50. PubMed Abstract
| Publisher Full Text
| Free Full Text
49.
Nakashima M, Miyajima M, Sugano H, et al.:
The somatic GNAQ mutation c.548G>A (p.R183Q) is consistently found in Sturge-Weber syndrome.
J Hum Genet.
2014; 59(12): 691–3. PubMed Abstract
| Publisher Full Text
53.
Zeng H, Zhao D, Yang S, et al.:
Heterotrimeric G alpha q/G alpha 11 proteins function upstream of vascular endothelial growth factor (VEGF) receptor-2 (KDR) phosphorylation in vascular permeability factor/VEGF signaling.
J Biol Chem.
2003; 278(23): 20738–45. PubMed Abstract
| Publisher Full Text
54.
Wettschureck N, Offermanns S:
Mammalian G proteins and their cell type specific functions.
Physiol Rev.
2005; 85(4): 1159–204. PubMed Abstract
| Publisher Full Text
55.
Hubbard KB, Hepler JR:
Cell signalling diversity of the Gqalpha family of heterotrimeric G proteins.
Cell Signal.
2006; 18(2): 135–50. PubMed Abstract
| Publisher Full Text
56.
Morris PN, Dunmore BJ, Tadros A, et al.:
Functional analysis of a mutant form of the receptor tyrosine kinase Tie2 causing venous malformations.
J Mol Med (Berl).
2005; 83(1): 58–63. PubMed Abstract
| Publisher Full Text
57.
Osborn AJ, Dickie P, Neilson DE, et al.:
Activating PIK3CA alleles and lymphangiogenic phenotype of lymphatic endothelial cells isolated from lymphatic malformations.
Hum Mol Genet.
2015; 24(4): 926–38. PubMed Abstract
| Publisher Full Text
59.
Luks VL, Kamitaki N, Vivero MP, et al.:
Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA.
J Pediatr.
2015; 166(4): 1048–54.e1–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
61.
Yuksekkaya H, Ozbek O, Keser M, et al.:
Blue rubber bleb nevus syndrome: successful treatment with sirolimus.
Pediatrics.
2012; 129(4): e1080–e1084. PubMed Abstract
| Publisher Full Text
63.
Boscolo E, Limaye N, Huang L, et al.:
Rapamycin improves TIE2-mutated venous malformation in murine model and human subjects.
J Clin Invest.
2015; 125(9): 3491–504. PubMed Abstract
| Publisher Full Text
| Free Full Text
65.
Lackner H, Karastaneva A, Schwinger W, et al.:
Sirolimus for the treatment of children with various complicated vascular anomalies.
Eur J Pediatr.
2015; 174(12): 1579–84. PubMed Abstract
| Publisher Full Text
67.
Kwan DH, Yung LY, Ye RD, et al.:
Activation of Ras-dependent signaling pathways by G14-coupled receptors requires the adaptor protein TPR1.
J Cell Biochem.
2012; 113(11): 3486–97. PubMed Abstract
| Publisher Full Text
68.
Lim YH, Douglas SR, Ko CJ, et al.:
Somatic Activating RAS Mutations Cause Vascular Tumors Including Pyogenic Granuloma.
J Invest Dermatol.
2015; 135(6): 1698–700. PubMed Abstract
| Publisher Full Text
| Free Full Text
69.
Bos JL:
ras oncogenes in human cancer: a review.
Cancer Res.
1989; 49(17): 4682–9. PubMed Abstract
71.
Trombetta D, Magnusson L, von Steyern FV, et al.:
Translocation t(7;19)(q22;q13)−a recurrent chromosome aberration in pseudomyogenic hemangioendothelioma?
Cancer Genet.
2011; 204(4): 211–5. PubMed Abstract
| Publisher Full Text
72.
Walther C, Tayebwa J, Lilljebjörn H, et al.:
A novel SERPINE1-FOSB fusion gene results in transcriptional up-regulation of FOSB in pseudomyogenic haemangioendothelioma.
J Pathol.
2014; 232(5): 534–40. PubMed Abstract
| Publisher Full Text
74.
Declerck PJ, Gils A:
Three decades of research on plasminogen activator inhibitor-1: a multifaceted serpin.
Semin Thromb Hemost.
2013; 39(4): 356–64. PubMed Abstract
| Publisher Full Text
75.
Antonescu CR, Chen HW, Zhang L, et al.:
ZFP36-FOSB fusion defines a subset of epithelioid hemangioma with atypical features.
Genes Chromosomes Cancer.
2014; 53(11): 951–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
80.
Wine Lee L, Goff KL, Lam JM, et al.:
Treatment of pediatric pyogenic granulomas using β-adrenergic receptor antagonists.
Pediatr Dermatol.
2014; 31(2): 203–7. PubMed Abstract
| Publisher Full Text
1
Department of Dermatology, Yale University School of Medicine, New Haven, CT, USA 2
Department of Pathology, Yale University School of Medicine, New Haven, CT, USA 3
Department of Genetics, Yale University School of Medicine, New Haven, CT, USA
The work for this report was supported by the Leon Rosenberg, M.D., Medical Student Research Fund in Genetics and the Jane Danowski Weiss Family Foundation Fellowship at the Yale University School of Medicine.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Cheraghlou S, Lim Y and Choate K. Genetic investigation of childhood vascular tumor biology reveals pathways for therapeutic intervention [version 1; peer review: 2 approved]. F1000Research 2019, 8(F1000 Faculty Rev):590 (https://doi.org/10.12688/f1000research.16160.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)