Keywords
RNA-Seq data, Logarithmic transformation, Biomarker, Outliers and Robustness
RNA-Seq data, Logarithmic transformation, Biomarker, Outliers and Robustness
One of the major objectives of researchers is to identify biomarkers from RNA-Seq data that are differentially expressed (DE) between two or more experimental conditions. Microarrays have been extensively used during the past few decades to perform this task. But after reducing the cost of sequencing, biomarker identification using RNA-seq data has emerged as an alternative choice to microarrays1,2. RNA-seq uses next generation sequencing technology to produce a vast amount of data. Curse of dimensionality is a common problem for analyzing RNA-Seq data, which means a "large p, small n" problem. Hence, dimension reduction of the data matrix is a primary objective for further downstream analysis. Identification of biomarkers is one of the dimensionality reduction approaches.
RNA-seq count data inherently follow a Poisson or negative binomial (NB) distribution rather than normal distribution like microarray data. Numerous statistical methods have been developed to identify biomarkers from RNA-seq count data. The earliest method is DEGseq, which is based on Poisson distribution. This method suffers from overdispersion and therefore Poisson distribution-based methods are not suitable for RNA-seq data3–6. To overcome this problem, NB distribution-based methods have been proposed. Some NB based methods are: baySeq, DESeq, DESeq2, EBSeq, edgeR, edgeR (robust) and NBPSeq7–11. However, most of the methods cannot estimate properly the gene-wise dispersion parameters and they also suffer from small sample sizes12. DESeq, DESeq2, edgeR, edgeR (robust) and NBPSeq incorporate information of all genes in their algorithms.
Despite the popularity of these statistical methods for identification of biomarker genes, they are sensitive to outliers and often produce lower accuracies in the presence of outliers. Outliers may arise in RNA-seq count data because there are several data generating stages from biological harvesting of RNA samples to counting of sequence read map data13. To mitigate this issue many algorithms use transformation methods. There are several transformation methods for RNA-seq data: logarithmic transformation14, variance-stabilizing transformation (vst)6, TMM transformation15, regularized logarithm8 and variance modeling at the observation level (voom)16. These methods only reduce the low level outliers into reasonable spaces during parameter estimations; however they fail to reduce the influence of high level outliers with small sample sizes in the data matrix.
Consequently, most biomarker selection methods that use the aforesaid transformations, are sensitive to outliers or extreme values with small-sample sizes. Therefore, in this study, an attempt is made to propose an outlier detection and modification approach for RNA-seq data to improve the performance of the popular biomarker selection methods in the presence of outliers. To make our proposed method facilitate in RNA-seq data we transform the read count data into continuous data using regularize logarithmic transformation.
The article is organized as follows: Methods briefly describes the logarithmic transformation and formulation the proposed outlier detection and modification approach. In Results and Conclusions a broad simulation study and a real data study have been carried out.
Let ygik be the number of reads simulated from gth gene of kth replicates in the ith condition (g = 1, 2, . . ., G; i = 1,2; k =1, 2, . . . , ni); where ni is the number of replicated in condition i. G is the total number of gene. Then the negative bionomial-distribution is as follows:
In this negative binomial parameterization, E(ygik) = μgi and Var ( where, μgi is the mean of gth gene in ith condition and is the dispersion parameter. Now we want to test the following null hypothesis:
A gene will be declared as DE if H0 is rejected, otherwise it is equally expressed (EE).
Log-transformation is very useful in RNA-Seq data. The log-transformed data usually follow the normal distribution, which depends on the degree of skewness before transformation. As the RNA-Seq count data can be equal to zero, so we shift them by one before transforming them:
Log-transformed values have less extreme values (or outliers) than the untransformed data.
However, the log-transformed values reduces the influence of low level outlying observations; however this transformation fails to reduce the influence of high level outlying observations. Therefore, we propose the following outlier detection and modification rule.
Since the median and median absolute deviation is the robust measure of location and scale parameter, respectively, therefore, we used the median control chart as a measure of outlier detection. The proposed procedure is as follows:
1. We declare a gene as an outlying gene if it doesn’t fall into the interval [LCL, UCL]. Where LCL and UCL are the lower and upper control limit for median and they are defined by LCL= MEDg,(i)-3×MADg,(i) and UCL=MEDg,(i)+3×MADg,(i)]. Here, MEDg,(i)=median
(xgik; g =1, 2,…, G; k =1, 2,…, ni ; i=1,2) is the median of gth gene in ith condition, MADg,(i) = mediank=1,2,..,ni(|xgik − MEDg, (i)|) is the median absolute deviation.
2. Check the existence of outliers for each gene from each of the conditions (i=1,2), separately using step 1. If outliers exist, replace them by their respective group medians (MEDg,(i)).
3. Apply the anti-log transformation to obtain modified RNA-seq (MRS) count datasets.
4. Apply traditional statistical methods in MRS data to identify biomarker genes using the p-values adjusted by Benjamini-Hochberg method.
5. Obtain the functional annotations and KEGG pathways for detected biomarker genes.
In order to evaluate the performance of different biomarkers selection methods we considered the area under the receiver operating characteristic curve (ROC) curve. The ROC is created by plotting the true positive rate (TPR) against the false positive rate (FPR) for different cut-off points of a parameter. For a particular threshold each point on the ROC curve produces a TPR/FPR pair. The area under the ROC curve (AUC) is a performance measure which helps us to select an optimum method that can distinguish between two gene groups such as DE or EE well.
To investigate the performance of the proposed method in comparison with five popular methods as mentioned above, for both small-and-large-sample cases with 2 groups/conditions, we considered 100 datasets for both cases with sample sizes of n1=n2= 3 and 15, respectively. Each dataset for each case represented gene expression profiles for 1000 genes, each with n=(n1+n2) samples, where the read counts of each gene was generated using the negative-binomial distribution and this type of simulation study was also used in 11. The number of DE genes were set to 40 for each of the 100 datasets. We divided these 40 DE genes into two groups: 20, up-regulated DE genes and 20, down-regulated DE genes. To show the effect of outliers (extreme high counts) on the methods, we randomly selected 10% and 30% genes and for each of these genes, we selected a single sample randomly and multiplied the observed count of this sample with randomly selected factor between 5 and 10. This process was applied for each of the 100 datasets. We computed average values of different performance measures such as true positive rate (TPR), false positive rate (FPR) and AUC based on 40 estimated DE genes by five methods (edgeR, edgeR_robust, DESeq, DESeq2, and limma (voom)) for each of 100 original datasets and proposed MRS datasets.
We also considered a real RNA-seq mouse dataset17 to demonstrate the performance of the methods. This dataset consists of 36535 genes with 21 samples. This dataset was downloaded from ReCount website http://bowtie-bio.sourceforge.net/recount. It can also be downloaded from the GEO series accession number GSE26024. Among 21 samples, RNA-seq count expression collected from 10 C57BL/6J (B6) and 11 DBA/2J (D2) inbred mouse strains.
To demonstrate the performance of the proposed method, a comparison with five popular methods (edgeR, edgeR_robust, DESeq, DESeq2 and limma (voom)) was performed. We used both simulated and real RNA-seq count datasets. We used three R packages of other methods: edgeR, DESeq and limma. The performance measure area under the receiver operating characteristics curve (AUC) was computed for each of the methods using R package ROCR. All R packages are available in the comprehensive R archive network (cran) or Bioconductor.
Table 1 summarizes the average AUCs estimated by eight methods based on 100 simulated datasets using 4% DE genes in absence and presence of single outlier in each of 10% and 30% genes for both small-sample cases (n1=n2=3) and large-sample cases (n1=n2=15), respectively. In Table 1, the results without and within the brackets (.) indicate the estimated AUCs by the five methods using the original RNA-seq datasets and proposed MRS datasets. From Table 1 we observed that in absence of outliers, five methods (edgeR, edgeR_robust, DESeq, DESeq2 and limma (voom)) produced almost similar results using the original RNA-seq datasets and proposed MRS datasets, for both small-and-large-sample cases. However, in the presence of outliers, the performance of these methods has significantly increased using the proposed MRS datasets for both cases. For example, in the presence of 10% outliers, edgeR and DESeq produce AUCs 0.829 and 0.818, respectively for small-sample case. Whereas for the same condition these two methods produce AUCs 0.842 and 0.838, respectively using our proposed MRS datasets. Figure 1a and b and Figure 1c and d show the boxplot of AUCs based on 100 simulated datasets by each of the methods in absence and presence of outliers for small-and large-sample cases, respectively. The left and right-side panels in this figure indicate the boxplot of estimated AUCs using original RNA-seq datasets and proposed MRS datasets. Similar results were found from these boxplots, as in Table 1.
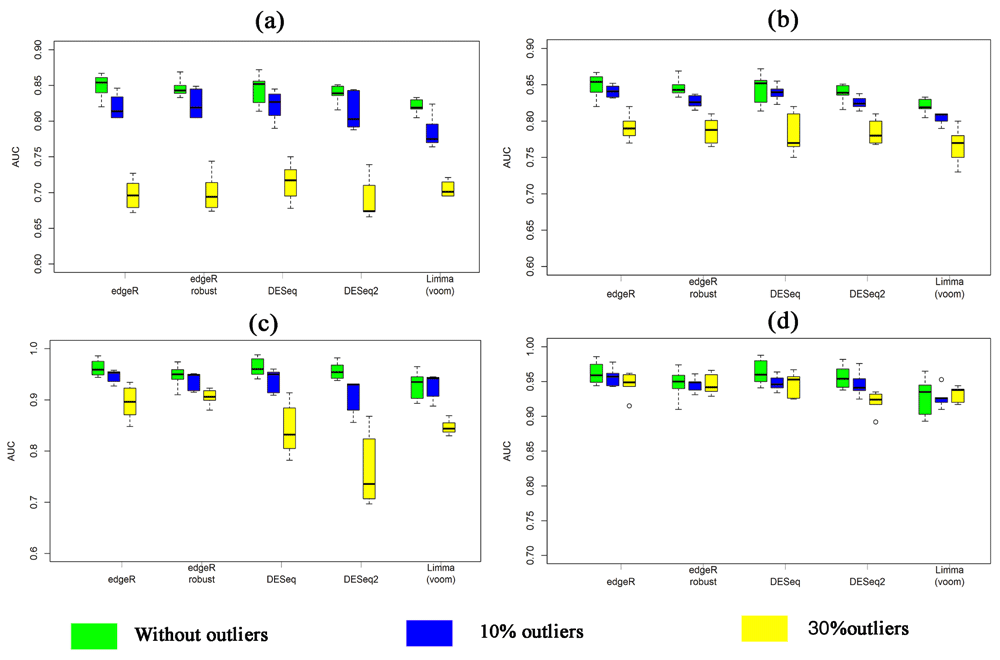
(a–b) for small-sample case (n1=n2= 3) and (c–d) for large-sample case (n1=n2= 15).
After filtering we retain 11,474 genes. To investigate the performance of the proposed method, we employ three methods (edgeR, DEseq and limma) for detection of the biomarkers between the two mouse strains. Figure 2a and b represents the Venn diagram of estimated DE genes by edgeR, DESeq and Limma using the original and MRS dataset, respectively. From Figure 2a, we revealed that edgeR and Limma performed better than DESeq by sharing more genes (414). We also noticed that there are 1925 overlapping DE genes between these methods. To investigate the performance of the proposed outlier detection and modification approach in this dataset, we first detect and modify the outliers (if any) to get the MRS dataset. We detected 200 outliers in this dataset using the proposed outlier detection rule. The Venn diagram in Figure 2b represents the results of these three methods using the proposed MRS dataset. From this figure we can clearly observe that there are 1956 overlapping genes detected by these methods. Among these genes there are 18 genes that are declared as outliers by the proposed method and those were not detected as DE genes using the original mouse dataset.
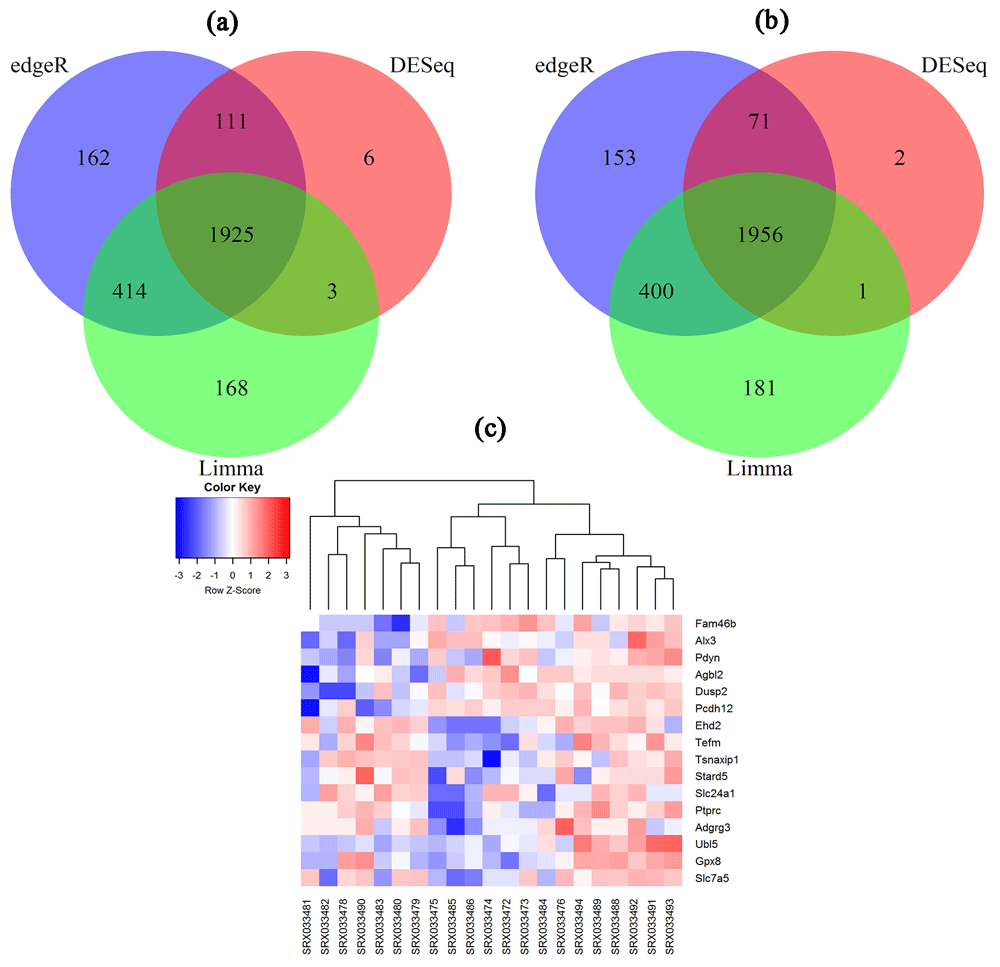
Venn diagram of DEGs detected by (a) the edgeR, DESeq and Limma in the original mouse dataset or by (b) the edgeR, DESeq and Limma in the modified mouse dataset using the proposed method. (c) Heatmap of 16 outlying DEGs detected by the proposed method.
Furthermore, we performed the gene overexpression analyses through Database for Annotation, Visualization and Integrated Discovery (DAVID)18 to explore the biological process (BF) categories and pathway annotations of the 18 identified outlying DE genes. Out of 18 genes DAVID identified 16 genes. A heatmap is created for these 16 outlying DE genes in Figure 2c. The heatmap correctly clusters the samples between C57BL/6J (B6) and 11 DBA/2J (D2) using these genes. Among the 16 genes, 8 upregulated (Fam46b, Alx3, Dusp2, Pdyn, Agbl2, Pcdh12, Ubl5, Gpx8) and 8 downregulated (Stard5, Ptprc, Slc7a5, Slc24a1, Ehd2, Adgrg3, Tefm, Tsnaxip1) DE genes are identified. GO analysis results showed that upregulated DE genes are significantly enriched in protein side chain deglutamylation, protein deglutamylation and embryo development at BP level. Downregulated DEGs were enriched in negative regulation of CREB transcription factor activity, negative regulation of NIK/NF-kappaB signaling and sterol import at BP level (see extended data). KEGG analysis showed that the upregulated DEGs were mostly enriched in cocaine addiction, glutathione metabolism and thyroid hormone synthesis. The downregulated DEGs were enriched in phototransduction, primary immunodeficiency and central carbon metabolism in cancer (Table 2). We also constructed the protein-protein interaction (PPI) network around the proteins encoded by these 16 outlying genes using STRING database19. We considered confidence score 400 for selection these networks. Figure 3 and Figure 4 represent the PPI networks using the up-regulated and down-regulated DEGs, respectively. In addition, we explored miRNAs-target gene interactions from miRTarBase20 to identify miRNAs. The miRNAs-target gene interactions network is shown in Figure S1 in extended data.
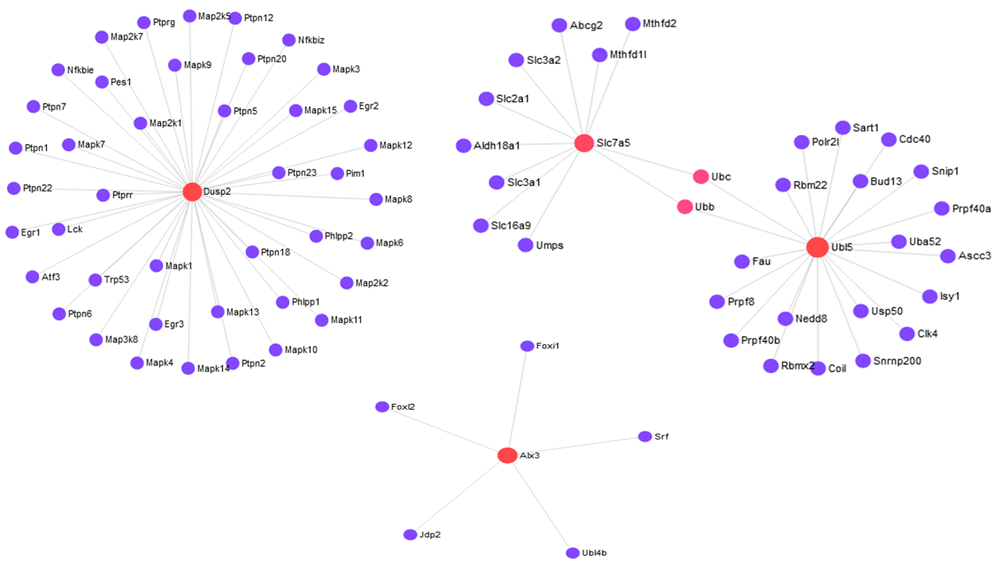
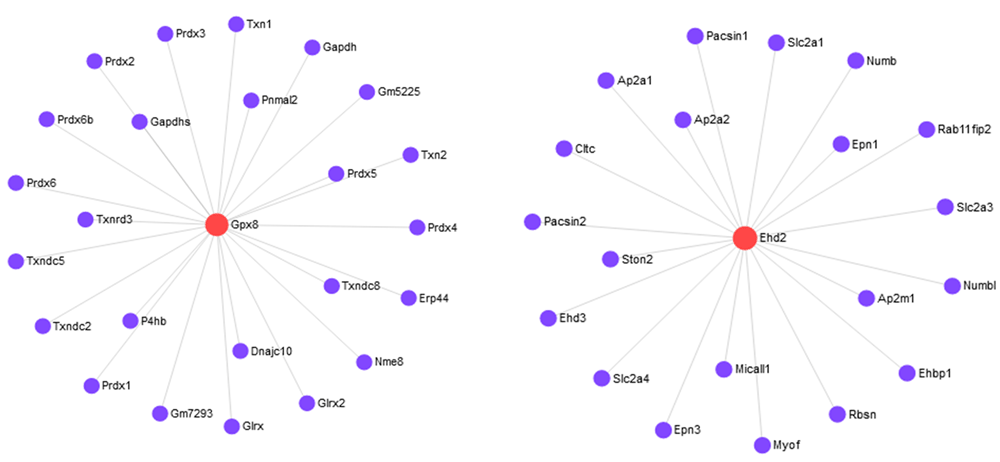
Biomarker identification under two or more conditions is an important task for elucidating the molecular basis of phenotypic variation. Next generation sequencing (RNA-seq) has become very popular and a competitive alternative to microarrays because of reducing the cost of sequencing and limitation of microarrays. A number of methods have been developed for detecting biomarkers from RNA-seq data. However, most of the methods are sensitive to outliers and produce misleading results in the presence of outliers. In this study, we have proposed an outlier detection and modification approach using the median control chart. From the simulation study in the presence of outliers we have observed that the performance of five biomarker selection methods are improved significantly when the datasets are modified by the proposed method, both for small-and large-sample cases. The proposed method also detected an additional 16 outlying genes from a real mouse dataset. From GO and KEGG pathway enrichment analysis, we have shown that these genes belong to some important pathways.
Simulated datasets available from: https://doi.org/10.5281/zenodo.221288121
Real dataset: The mouse dataset used in this study is publicly available at the NCBI GEO website: GSE26024.
Zenodo: Figure S1: miRNAs-target gene interactions using the outlying genes identified by the proposed method, http://doi.org/10.5281/zenodo.227992122
Zenodo: Table A1. Biological process categories for 16 genes, http://doi.org/10.5281/zenodo.228001223
The R code for the proposed method is available in https://github.com/snotjanu/OutMod-RnaSeq
Archived code: http://doi.org/10.5281/zenodo.227940524
License: MIT
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)