Keywords
RSV, methyltransferase, antiviral target, replication, broad spectrum, polymerase, methylation, MTase, innate immunity, homology modeling
RSV, methyltransferase, antiviral target, replication, broad spectrum, polymerase, methylation, MTase, innate immunity, homology modeling
The purpose of our opinion article is to draw a wider attention to the unmet medical need for treating HRSV infections and to highlight MTase as a potential target. In the past decade, targeting protein methyltransferases have led to many successful clinical candidates for cancer treatment (Philos Trans R Soc Lond B Biol Sci. 2018 Jun 5; 373(1748): 20170080). We also use this article to highlight key areas where drug discovery research needs to focus its attention in order to advance the field and aid in the identification of novel agents to treat HRSV infection.Our goal is not to be exhaustive but to make sure that the appropriate references are included so that the readers could be directed to those to find mechanistic details.As such we believe our article is timely and serves this intent.
In the revised version of the manuscript, we have updated the virus classification terminology, polymerase "regions" instead of "domains" and added 2 new references (Rima et al 2017 and Decroly et al 2011). Distinction in the capping mechanisms and Li Lab references (for HMPV work) have been added. As per the reviewer’s suggestion, we have added a figure (Figure 1) to illustrate the vRNA capping. We have mentioned that SAM is the methyl donor and SAH is a byproduct of MTase activity and in Figure 1 and removed redundancies.
Late elongation and polyadenylation reactions happen subsequent to methylation of viral RNA cap per Liuzzi et al 2005. Hence it appears that inhibition of viral cap methylation leads to reduction in transcripts with late elongation and polyadenylation. Since viral MTase have multiple catalytic residues required for enzyme activity, mutation at multiple contact residues in the catalytic site might be needed to overcome antiviral activity of an inhibitor.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Human respiratory syncytial virus (HRSV) causes lower respiratory tract infections (bronchiolitis and pneumonia) in premature babies, young children, immuno-compromised adults and bone marrow transplant patients1,2. It is a significant risk factor for asthma, wheezing2 and progression of chronic obstructive pulmonary disease (COPD)3,4. However, the molecular pathogenesis of HRSV in asthma, wheezing and COPD is not clear. HRSV-associated infections result in significant disease and mortality. In children under 5-years of age, 33.4 million cases of HRSV-associated acute lower respiratory tract infections which result in 3.4 million hospitalizations and between 53,250 and 199,000 deaths, are reported annually worldwide5.
HRSV, now formally known as human orthopneumovirus, is an enveloped RNA virus with non-segmented negative sense RNA (nsNS) genome, belonging to the Pneumoviridae family6. There are two subtypes of HRSV, subtype -A and -B, circulating in the population. HRSV causes significant health problems but at present there is no effective treatment or vaccine. The U. S. Food and Drug Administration (FDA) has approved palivizumab7, for prophylactic therapy in premature infants with less than 29-weeks of gestation and children with congenital lung or heart disease. Ribavirin, a broad-spectrum antiviral agent is the only FDA approved drug for the treatment of severe HRSV disease8. However, the use of ribavirin is limited due to the potential side effects, high cost, difficulty in administration and lack of demonstrated benefit in decreasing hospitalization and mortality9. Currently, the few drug candidates in clinical development (GS-5806, ALS-8176, EDP-938) are directed towards a limited number of viral targets (HRSV F protein, polymerase and N protein respectively)10–12. Thus, novel targets in HRSV replication are needed to address the clinically unmet medical need13.
HRSV produces mRNAs, which are co-transcriptionally capped and polyadenylated14–16. Viral mRNA methyltransferases (MTases) catalyze the transfer of methyl groups from SAM (S-adenosylmethionine; methyl donor) to viral mRNA caps comprised of a guanosine nucleotide linked via a unique 5’-5’ linkage17. These processes of capping and methylation are distinct in different viruses, even though the end products are chemically equivalent and contribute to mRNA stability and translation efficiency18. Previous analysis of the L-protein sequence of non-segmented negative strand (nsNS) RNA viruses revealed the presence of 6-conserved regions (I-VI)19. These regions were implicated in viral genome replication, transcription, cap methylation and 3’ polyadenylation19,20. Fearns and Deval have graphically illustrated these regions using the VSV RNA polymerase as an example13. Using computational analyses of L protein of the Mononegavirales family viruses alongside site-directed mutagenesis, the MTase domain has been previously mapped to region VI, with a putative K-D-K-E catalytic tetrad and a glycine-rich motif (GxGxG) (SAM binding site)20,21. A hallmark of the SAM-dependent MTase superfamily is the conserved sequences (segregated into motifs I-X) responsible for two-conserved functions- SAM binding and MTase catalytic reaction. Motifs I, III and IV are shown to be involved in SAM binding, whereas motifs IV, VI, VIII and X play a major role in the catalytic reaction19. Ribose 2’-O MTase is typically shown to have K-D-K-E tetrad residues at its core and these residues participate in the catalytic methylation reaction.
Viral mRNA cap methylation process for vesicular stomatitis virus (VSV) has been well understood (a prototype for nsNS group of viruses) model (graphically illustrated in Figure 113). As per this model, viral mRNA capping process is thought to involve three different enzymatic steps (1) GTPase (2) GDP polyribonucleotidyltransferase (PRNTase) and 3) MTase. This viral mRNA cap methylation model, based on published literature, has revealed some unique features that are distinct from the host mechanism20. These include (1) dual specificity of MTase activity on both the N-7 guanosine and 2’-O ribose positions encoded in a single conserved region (CR VI) of L-protein; (2) sharing of the same binding site for S-adenosylmethionine (SAM), that acts as the methyl donor22–24 (3) 2’-O methylation preceding and facilitating G-N-7 methylation25, and (4) requirement of cis-elements in viral RNA for cap methylation. Mechanisms involved in mRNA capping functions of HRSV L-protein are not clearly understood. Ogino and coworkers26 have shown that the mRNA cap for VSV and other rhabdoviruses is added by a PRNTase activity, rather than by guanylylation. Due to the similarities within their capping domains, it is likely that HRSV uses the same mechanism as the rhabdoviruses, although this has not yet been experimentally demonstrated.
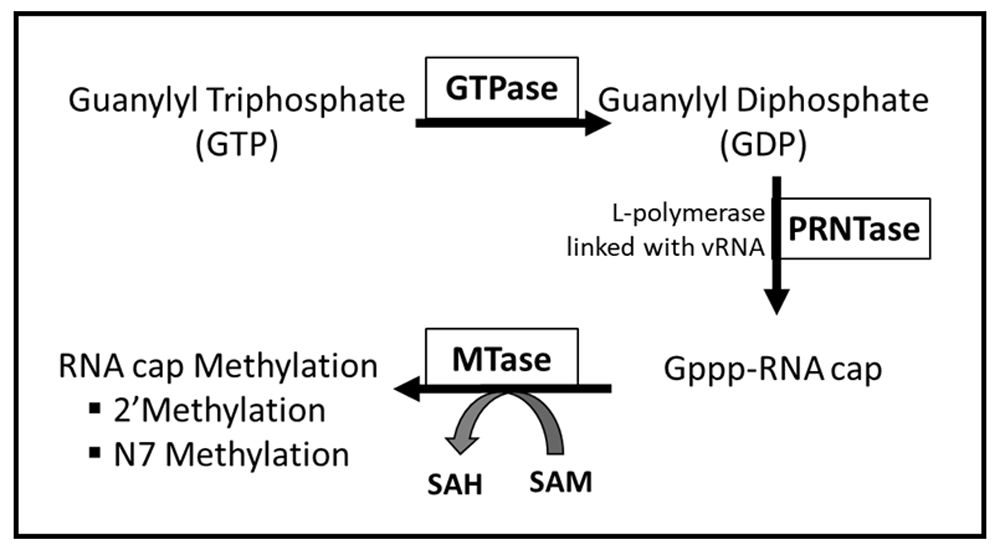
A proposed model for HRSV MTase in the methylation and capping of RNA based on similarities to VSV and other rhabdoviruses: GTPase hydrolyzes Gulanylyl Triphosphate (GTP) to Guanylyl Diphosphate (GDP). GDP polyribonucleotidyltransferase (PRNTase) facilitates the covalent linkage of the 5’-end of viral RNA with viral polymerase (L–protein) after removal of pyrophosphate. GpppRNA:cap product is formed after GDP attacks the phosphodiester bond between L-protein and RNA. Viral MTase methylates the viral RNA cap at 2’-position of ribose of the first nucleotide and N7 of the first guanine in viral RNA cap in a sequential manner. For the methylation reaction, MTase uses S-adenosyl methionine (SAM) as substrate, with S-adenosyl-L-homocysteine (SAH) as a byproduct.
Alignment of the L-protein sequences from different members of the Pneumoviridae and other nsNS RNA viruses has demonstrated conserved residues in the MTase domains21. As previously reported, conserved motifs within the HRSV MTase domain could be predicted between amino acid sequences F1821-N2025, with catalytic tetrad at K1831, D1936, K1973, E2004 and a putative SAM-binding GxGxG….D motif at positions G1853-E-G1855-A-G1857 and D1912 in HRSV L protein. MTase catalytic tetrad (K-D-K-E) and SAM binding domains (GxGxG…D) are conserved in CR VI of HRSV and VSV (Vesicular Stomatitis Virus), suggesting a similar mechanism of cap methylation26–28.
An X-ray crystal structure of HRSV MTase domain is not available in the literature. Previously, a 3.8 Å resolution structure of VSV L-protein and the methyl transferase domain using electron cryomicroscopy has demonstrated that the MTase contacts both the connector and the capping domains, without direct contact with the RNA dependent RNA polymerase (RdRp)29. However, the authors also predicted the substantial conformational change in the L protein following initial polymerization. The lack of a high resolution X-ray crystal structure of the HRSV MTase is a major caveat for a structure based drug design effort.
In the absence of an X-ray crystal structure, homology modeling provides an alternative approach to model the protein structure using the crystal structure of related protein(s) where significant sequence identity/similarity exists. Sequence alignment of HRSV polymerase suggested an overall low protein sequence similarity among the members of different nsNS viruses. Similarly, low sequence homology (8–13%) was observed between HRSV MTase domain and those for which X-ray structures were available namely dengue virus NS5, vaccinia virus VP39 and E. coli RrmJ that precluded the building of a homology model of HRSV MTase. Fortunately, crystal structure of human metapneumovirus (HMPV) methyltransferase were reported recently30. HMPV is a pneumovirus of the Pneumovirinae subfamily closely related to respiratory syncytial virus (HRSV). The MTase domains of these viruses have about 35% sequence identity and close to 60% sequence similarity allowing homology-based modeling of the RSV MTase domain. The suitability of crystal structure of human metapneumovirus (HMPV) methyltransferase for homology modeling of HRSV MTase and structure-based drug discovery needs to be further explored.
Recombinant VSVs (a prototype of nsNS RNA viruses) with point mutations in the methyltransferase catalytic site (rVSV-K1651A, -D1762A, and -E1833Q) were reported to be defective in cap methylation and demonstrated reduced growth in cell culture and mice31. Though SAM binding site point mutations (rVSV-G1670A, G1672A, G1674A and G4A), were attenuated in vitro, low level virulence was still observed in vivo20. In contrast, mutations in the SAM binding site of L-protein in recombinant flaviviruses and metapneumoviruses attenuated viral replication in cell culture and animal models (cotton rats and turkeys)27,28, supporting the importance of MTase for viral replication and virulence32.
Previously, HRSV transcription was shown to be independent of cap methylation, where S-adenosyl-L-homocysteine (SAH), a byproduct of MTase activity, did not affect HRSV transcription despite SAM-dependent inhibition of methylation33. However, this observation was based only upon in vitro transcription using infected cell extracts without evaluation of the quality/stability of the HRSV transcripts. For VSV (a prototype nsNS RNA virus), SAH was shown to affect the quality of mRNA (aberrant polyadenylation) without an apparent effect on transcription34,35. Thus, evaluation of the quality of transcribed viral mRNAs in the presence of SAH might reveal a previously uncharacterized effect on HRSV MTase activity. The HRSV minigenome assay provides an opportunity to address this possibility, as it is performed in cells with measurement of both transcription and translation36. Based on the model suggested for HRSV transcription, methylation of viral RNA cap occurs before late elongation and poly-adenylation16. Hence, it is possible that inhibition of viral cap methylation might lead to reduction in late elongation and polyadenylation of viral RNAs. However, additional studies are needed to confirm this hypothesis for HRSV MTase.
Recent reports suggest that amino acid substitutions in the conserved SAM binding site and MTase domain of metapneumoviruses result in defective mRNA cap methylations and attenuate viral replication in vivo27,28. Since metapneumoviruses also belong to the Pneumovirinae subfamily as HRSV, it is possible that inhibition of HRSV MTase and SAM binding functions will negatively affect viral mRNA transcription and consequently, viral replication.
Several small molecule inhibitors of viral MTase such as Sinefungin (SIN) and S-adenosyl-L-homocysteine (SAH) derivatives have been reported. SIN, a natural SAM-analog and a potent inhibitor of MTase, shows antiviral activity against VSV, Newcastle disease and vaccinia viruses37. Similarly, derivatives of SAH, a byproduct of mRNA cap methylation, have shown selective inhibition of MTase of dengue virus38. Key residues in HRSV MTase catalytic motif and SAM binding domain seem to be conserved between different subtypes A and B of HRSV, pneumoviruses and negative sense non-segmented viruses except Bornaviridae family members21,39. Such a conserved sequence could provide a basis for structure-based design for pan-antiviral inhibitors targeting viral MTase28. The recently established X-ray structure of the MTase domain of HMPV might allow us to build a homology model for HRSV MTase30. This new information could facilitate structure-guided drug design.
The 2’O methylation of viral RNA is reported to be important for viral evasion of host innate immunity. The interferon-induced proteins with tetratricopeptide repeats (IFIT) are a part of the innate immune response needed to defend against viruses, recognize unmethylated mRNAs as “non-self” and target them for degradation, thus underscoring the importance of mRNA capping in replication of West Nile virus (WNV), Poxviruses, Coronaviruses, and HMPV28,32. Although additional studies are needed to confirm this phenomenon for HRSV, one could envision a potential “double whammy” effect of HRSV MTase inhibition on viral replication; on one hand the inhibition of MTase activity may attenuate or block viral transcription and replication whilst the generation of unmethylated viral mRNA caps will result in its degradation by the cellular innate immune response machinery. Since viral MTase have multiple (4) catalytic residues required for enzyme activity, mutation at multiple contact residues in the catalytic site might be needed to overcome antiviral activity of an inhibitor. Moreover, targeting HRSV MTase at the SAM binding site might offer a high barrier to drug resistance, as mutations at key inhibitor-contact residues required for SAM-binding would attenuate interactions with SAM and result in reduced catalytic efficiency, eventually leading to poor replicative fitness. Thus, targeting HRSV MTase for antiviral activity might offer multiple benefits. Mechanistic distinctions between nsNS viral MTases (using VSV as a prototype) in comparison to host MTases could enable the discovery of antivirals that selectively target viral MTases but spare host MTases thus minimizing potential toxicity25.
The high degree of sequence conservation of the HRSV MTase catalytic residues and the fundamental differences between viral and host capping mechanisms combined with the potential for the restoration of innate immune response that could specifically degrade viral mRNAs makes HRSV MTase a logical target for HRSV drug discovery efforts. Due to the sequence similarity of HRSV MTase with other members of the paramyxoviridae family, it is likely that an HRSV MTase inhibitor also have activity against paramyxoviruses. Such an antiviral spectrum might be added value for empirical treatment, especially due to unavailability/delay of virus-specific diagnostics and short time to treatment initiation. HRSV MTase inhibitors should be counter screened against viral panels to determine the antiviral specificity as is the norm for antiviral drug discovery efforts during lead optimization.
No data are associated with this article.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)