Keywords
Chlamydia, pathogenesis, genetics, cell-cell junctions, organoids, infection models
Chlamydia, pathogenesis, genetics, cell-cell junctions, organoids, infection models
The order Chlamydiae are obligate intracellular pathogens of eukaryotic cells. These bacteria have reduced genomes and display biphasic developmental stages that alternate between distinct extracellular and intracellular forms1,2. Eleven pathogenic Chlamydia species infect vertebrate animals and display tissue-specific tropism3. The elementary body (EB) is the environmentally stable form of the pathogen that binds and invades target cells. The EB then transitions into the larger intracellular reticulate body (RB) form. RBs replicate and secrete proteins across the parasitophorous membrane-bound vacuole (“inclusion”) to modulate multiple host cellular functions that benefit the bacterium4.
All Chlamydiae encode a type III secretion (T3S) system to deliver a defined cohort of bacterial proteins (“T3S effectors”) directly into the host cell5. The Chlamydia trachomatis EB T3S effectors modify the cytoskeleton and stimulate bacterial uptake into a membrane-bound vacuole that is rapidly segregated from degradative trafficking pathways (Figure 1A)6,7. A subset of T3S effectors are inserted into the inclusion membrane. These inclusion membrane proteins (Incs), which are secreted throughout the infectious life cycle, are diverse (~5% of the total coding potential of C. trachomatis) and their molecular function is just beginning to be understood8. For instance, Incs co-opt the microtubule motor protein dynein to transport nascent inclusions along microtubules toward the centrosome (Figure 1A)9. Along the way, the inclusion membrane is modified by lipid kinases, which may be important for evasion of endolysosomal compartments10. Some Incs contain SNARE (soluble N-ethylmaleimide–sensitive factor attachment protein receptor)-like domains that coordinate fusion between inclusions and other membrane vesicles11, whereas others promote the recruitment of the endoplasmic reticulum and Golgi complex to the vicinity of the inclusion, possibly to intercept lipid-rich vesicles to support Chlamydia replication (Figure 1A)12–14. As the inclusion matures, it is increasingly encased by a network of F-actin, microtubules, intermediate filaments, and septins, which help confine the bacteria within the inclusion and limit recognition of bacterial products by innate immune sensors15–17.
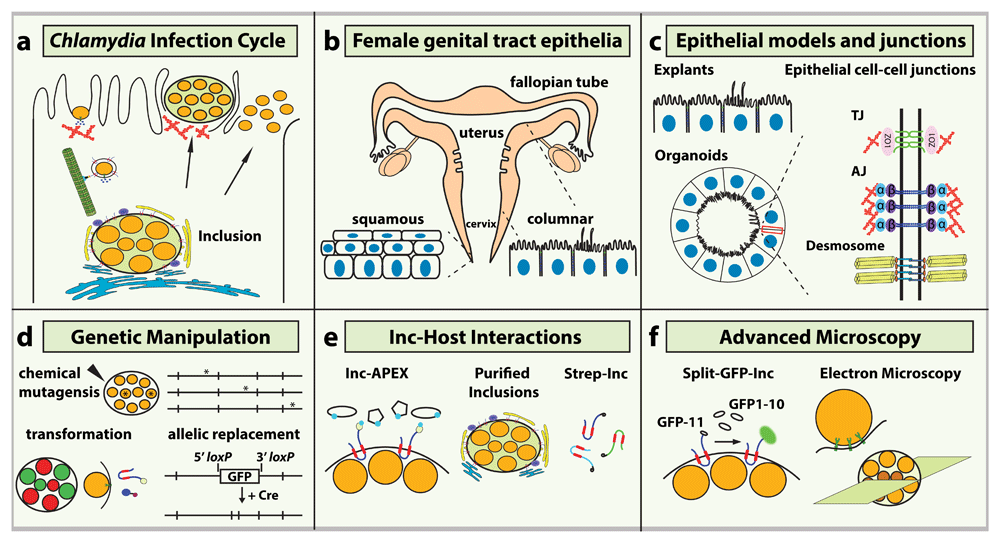
(a) The Chlamydia trachomatis infection cycle. The elementary body (EB) form of the bacteria remodels actin filaments (red) during entry and traffics along microtubules (green) to the perinuclear region. Inclusion membrane proteins (Incs) recruit the Golgi complex (yellow) and endoplasmic reticulum (blue). At the end of the intracellular cycle, the inclusion exits via actin-dependent extrusion or cell lysis. (b) Anatomy of the female genital tract and epithelial cell organization in the lower and upper tract. (c) New epithelial model systems (left) and schematic of polarized columnar epithelial cell–cell junctions (right). Tight junction (TJ) and adherens junction (AJ) complexes recruit adaptor proteins that connect to the actin cytoskeleton (red); desmosomes interact with intermediate filaments (yellow). (d) New genetic tools for C. trachomatis include chemical mutagenesis and whole-genome sequencing to identify mutations and plasmid transformation to generate fluorescent reporter strains, tagged effectors, and targeted gene disruption via allelic replacement. (e) New proteomic-based strategies to identify host proteins that interact with Chlamydia Incs. Incs tagged with the enzyme ascorbate peroxidase (APEX) (left) can ligate biotin-phenol on host proteins in close proximity. Purified inclusions (middle) and Strep-tagged Incs (right) were used to identify host proteins recruited to the inclusion and Inc–host protein interactions, respectively. (f) Summary of advanced microscopy approaches to visualize Chlamydia effector localization using the Split-green fluorescent protein (Split-GFP) system (left), the structure of the T3S apparatus in contact with the plasma membrane (middle), and reticulate body (RB)-to-EB conversion (right). ZO-1, zona occludens 1.
Mid-stage through the infectious cycle, RBs transition back to EBs such that at the end of the cycle the infectious bacteria are released either by an actin-dependent extrusion process whereby the intact inclusion is exocytosed from the cell or by lysis of the host cell which requires the cleavage of cytoskeletal elements and nuclear rupture18,19.
Much of our understanding of the cell biology of how Chlamydia interacts with target cells and the molecular mechanisms it uses to manipulate cellular processes is based on observations made in infected cancer cell lines, which in addition to being metabolically and genetically adapted for proliferation, lack positional cues that are available only in the context of tissues. For example, polarized columnar epithelial cells, the in vivo target of C. trachomatis, display a spatial organization of organelles and cell signaling pathways that intimately intertwine stable cell–cell junctions to epithelial function and proliferation (Figure 1B, C). Such structures and signaling networks are not present—or properly wired—in common cell lines used in Chlamydia research. Fortunately, more sophisticated infection models, coupled with the increasing ability to genetically manipulate Chlamydia, now provide a renewed tool kit to better understand how these pathogens interact with their intact animal hosts.
The greatest single advance in Chlamydia biology over the past decade has been the development of methods to perform genetic analysis of C. trachomatis mutants and increasingly robust molecular genetic tools to transform Chlamydia with recombinant DNA (Figure 1D)20–22. As a result, it is now possible to perform targeted gene inactivation and plasmid-based complementation of chromosomal mutations22–25. The ability to express exogenous proteins, epitope tags, and fluorescent and other reporter proteins in Chlamydia has also expanded the repertoire of possible technologies that can be applied to study the Chlamydia–host interface26,27. For instance, Incs fused to enzymes that enable the biotinylation of proteins identified host factors that are in proximity to the inclusion membrane by affinity capture of biotinylated proteins coupled with tandem mass spectrometry (Figure 1E)28,29.
Two recent studies using quantitative proteomics identified host proteins that interact with Incs and host proteins that are preferentially recruited to the inclusion (Figure 1E). In one study, intact inclusions were isolated and all associated host proteins were identified and quantified by stable isotope labeling by/with amino acids in cell culture (SILAC)-based mass spectrometry30. In parallel, a large-scale study identified previously unknown host-binding partners for nearly two thirds of the predicted Incs. Using affinity purification of Strep-tagged Incs coupled to quantitative mass spectroscopy, a second study provided a blueprint for Inc–host interactions31. The different but complementary approaches presented new clues as to the potential mechanisms used by Chlamydia to subvert various aspects of host cell biology.
The advances in genetic and biochemical approaches have been further complemented with high-resolution microscopy (Figure 1F). The localization and dynamics of Chlamydia T3S effectors can now be visualized live by using the split-green fluorescent protein (split-GFP) system32, an approach that relies on fusing the effector with the 16–amino acid GFP-11 β-strand and infecting cells expressing the GFP1-10 β-strands. Upon secretion and complementation, the GFP β-barrel properly folds and fluoresces (Figure 1F)33. Furthermore, the application of cryo-electron microscopy revealed with unprecedented resolution that the Chlamydia T3S apparatus changes shape and polarizes toward host cell membrane34. Similarly, applied serial block-face scanning electron microscopy temporally characterized the process of RB-to-EB conversion during the infection cycle (Figure 1F)35.
Urogenital serovars of C. trachomatis target the mucosal epithelium. In the female genital tract, the infection typically begins in the endocervix before ascending to the endometrium and fallopian tubes36. These tissues are comprised largely of polarized columnar epithelial cells with a rich diversity in form and function which is not readily recapitulated in two-dimensional culture settings37. Thus, new in vitro models that more accurately reconstruct the organization and complexity of the genital tract are essential to better understand the full impact of Chlamydia infection on the epithelial physiology.
Wyrick and colleagues first described key differences in Chlamydia growth in polarized epithelial cells38. Human endometrial epithelial cancer cells polarized on collagen-coated microcarrier beads significantly enhanced the growth of C. trachomatis serovar E, independent of EB attachment efficiency, compared with non-polarized cells38. Infections in polarized enterocytes show that the inclusion preferentially captures lipid-rich exocytic vesicles that are specifically trafficked toward the basolateral membrane, suggesting that Chlamydia has adapted to grow in a polarized environment39. More recently, a human endocervical epithelial cell line (A2EN cells) derived from a healthy patient sample has been shown to polarize, secrete mucin, and express pro-inflammatory cytokines during infection40,41.
Meyer and colleagues pioneered the use of ex vivo organotypic cultures and a novel fallopian tube organoid (FTO) model to investigate C. trachomatis infection in primary human epithelia42,43. Partially dissected tissue from the ectocervix and fallopian tube—representing the lower and upper genital tracts, respectively—has been cultured ex vivo and infected with Chlamydia42,43. These models were recently simplified by culturing isolated tubal epithelial cells in a three-dimensional matrix, generating self-renewing FTOs (Figure 1C)44. FTOs consist of secretory and ciliated epithelia, the two most common epithelial cell types in the fallopian tube, and more accurately recapitulate fallopian tube epithelial architecture. Acute infection in FTOs produced a strong inflammatory response while long-term and chronic infection increased epithelial stemness and proliferation and altered the methylation status of genes associated with aging. The latter results provide new insights into the long-term effects of Chlamydia infection on epithelial tissue homeostasis and the correlation between infection and cell proliferation45.
Ultrastructural analysis of the lower female genital tract shows squamous ectocervical epithelia and columnar endocervical epithelia, which display differential organization of cell–cell junctions37. The endometrium resembles columnar epithelia of the endocervix with increased epithelial diversity that includes ciliated and secretory cells and hormonally responsive and crypt-like glandular epithelia (Figure 1B)46.
Ultimately, the protective function of the epithelium is accomplished through the formation of intercellular junctions, molecular complexes that link up with the cytoskeleton to reinforce epithelial integrity. However, unlike other epithelial tissues, the endometrium exhibits remarkable changes during hormonal cycles and pregnancy, directly altering the expression profile of cell–cell junction proteins, their organization, dynamics, and barrier function46,47. In columnar epithelia, the three main classes of intercellular junctions are the tight junctions (TJs), adherens junctions (AJs), and desmosomes (Figure 1C)48. Although their function as molecular linkages has long been appreciated, newer studies have uncovered critical roles in transducing cell signaling pathways during tissue damage, repair, and pathogenic infection49.
At the apex of the lateral cell membrane, TJs maintain a fence that physically separates the apical and basolateral membranes while gating the flux of ions and solutes through the paracellular space50. Three families of transmembrane proteins—claudins, occludins, and junctional adhesion molecules (JAMs)—form homotypic interactions between adjacent cells. Adaptor proteins—that is, zona occludens 1–3 (ZO-1–3)—bind to their cytoplasmic tails and scaffold the recruitment of the polarity complex, which specifies apical membrane identity, and actin and microtubules to regulate TJ dynamics and stability50.
AJs along the basolateral membrane are formed by homotypic interaction between cadherins, calcium-dependent transmembrane proteins51. The adaptor proteins α- and β-catenin bind to cadherins and the actin cytoskeleton to reinforce AJ stability52. AJ components, including β-catenin and others such as YES-associated protein (YAP), also function as transcription factors but are excluded from the nucleus through their association with stable AJs53. AJ assembly is initially mediated by nectins, calcium-independent adhesion molecules, that also bind to AJ and TJ adaptor proteins54,55. The related but structurally distinct desmosomes are composed of a second class of cadherins—desmogleins and desmocollins—that connect to intermediate filaments, such as keratins, rigid cytoskeletal elements that provide additional structural support. Indeed, these strong junctions are thought to resist mechanical stress and are essential for the maintenance of epithelial integrity56.
Many pathogenic viruses and bacteria have evolved diverse strategies to subvert cell–cell adhesions to gain entry into host cells or penetrate further into the underlying tissue57. For example, Listeria monocytogenes surface protein InlA binds to E-cadherin and the hepatitis C virus binds to claudin-1 to promote their internalization58,59. The enteropathogenic Escherichia coli T3S effector EspF disrupts TJs and leads to a loss of epithelial barrier function60. Importantly, these perturbations to cell–cell junctions can elicit an immune response as lumenal bacteria and toxins leak into the underlying tissue61.
Initial observations in tumor cell lines indicated that C. trachomatis infection can impact cell–cell junctions. Chlamydia infections disrupt N-cadherin–based AJs in HeLa cells. Gaps were observed between Chlamydia-infected cells and β-catenin relocalized to inclusions based on indirect immunofluorescence62. These basic observations in two-dimensional cell culture were detailed further in an ex vivo fallopian tube infection model. Infection with a C. trachomatis lymphogranuloma venereum (LGV) biovar disrupted epithelial polarity and cell–cell junction organization through a Wnt-driven paracrine signaling pathway42. The Wnt pathway consists of secreted glycoproteins that bind to the Frizzled and LRP5/6 receptors, activating downstream signals to regulate tissue organization through cell fate determination, polarity formation, and cell growth63. Activation of the canonical pathway disrupts the “destruction complex”, stabilizing the Wnt effector β-catenin and allowing its translocation to the nucleus, where it activates target genes63. In fallopian tube epithelia, β-catenin translocates to the inclusion and a component of the destruction complex, adenomatous polyposis coli (APC), shows more diffuse cytoplasmic localization. Notably, the localization of these Wnt effectors also changed in neighboring uninfected cells but was rescued upon the addition of Wnt inhibitors, suggesting that infection can alter tissue-level Wnt signaling programs42. More recently, pharmacological inhibition of Wnt signaling in endometrial epithelial cells resulted in smaller and aberrant inclusions and significantly reduced the production of EBs64.
Chlamydia pneumoniae, the human respiratory pathogen and causative agent of roughly 4 to 6% of community-acquired pneumonia65, also targets effectors of the Wnt pathway. The C. pneumoniae Inc Cpn1027 interacts with and may recruit the Wnt effectors Caprin2 and glycogen synthesis kinase 3 (GSK3) to the inclusion66. The C. trachomatis T3S effector TepP regulates GSK3β recruitment to nascent inclusions in endocervical epithelial cells10, but it is unclear whether these effectors regulate Wnt in the appropriate tissue context. In endothelial cells, C. pneumoniae infection also promotes vascular endothelial (VE)-cadherin phosphorylation on Y65867, a residue that can control AJ organization and barrier function68. The actin-binding protein EPS8, which is recruited to nascent inclusions during invasion and interacts with phosphorylated peptides derived from the T3S effector Tarp, also binds to VE-cadherin, alpha-catenin, and TJ components to control junction organization69–72. These results suggest that Chlamydia may secrete effectors that can target components of cell–cell junctions and cell signaling pathways that regulate junction organization.
Chlamydia muridarum, a mouse-adapted Chlamydia, also alters the composition of TJ proteins in mouse oviduct epithelial cells73. Infection with C. muridarum decreases the expression of ZO-1, claudin-1/2, occludin, and JAM-1 while upregulating the expression of claudin-3 and claudin-4. Accordingly, C. muridarum infection alters transepithelial electrical resistance, indicating that the barrier function of TJs is compromised. These phenotypes were exacerbated by the absence of the Toll-like receptor 3 (TLR3), which initiates double-strand RNA (dsRNA)-dependent type I interferon responses74. How TLR3 signaling is linked to TJ remodeling is unclear, but type III interferons were recently reported to strengthen epithelial barrier function during Salmonella infection75.
Mechanistically, it remains to be determined whether reprogramming of cell signaling pathways or the removal of adhesion components and recruitment to the inclusion or both underlie the disruption of cell–cell junctions. Nevertheless, Chlamydia targeting of cell–cell junctions may drive certain aspects of its pathogenesis, including infertility and ectopic pregnancies. During estrus, the endometrium undergoes extensive paracrine-driven remodeling of cell–cell junctions to permit blastocyst adherence and invasion into the epithelial tissue46. Thus, infection-mediated disruption of epithelial cell–cell junctions may impede implantation. Alternatively, Chlamydia infection is associated with elevated expression of the prokineticins receptor 2 (PROKR2), a G protein–coupled receptor that modulates TJ organization and paracellular permeability, which may prime tubal epithelia for implantation in fallopian tubes of patients with ectopic pregnancies76,77.
The remodeling of epithelial cell–cell junctions is a hallmark of both inflammation and cellular proliferation78. New studies indicate that multiple Chlamydia biovars can induce the transformation of epithelial cells to a more mesenchymal-like state by activating the epithelial-to-mesenchymal transition (EMT) program. EMT is essential for organogenesis and tissue repair during which epithelial cells adopt features of cells in the mesenchyme like fibroblasts, altering their polarity and the organization of the actin cytoskeleton and cell–cell adhesions79. Driven by growth factors (for example, transforming growth factor beta [TGF-β], hepatocyte growth factor, and epidermal growth factor) and hormones (estrogen), EMT can be aberrantly activated during tumorigenesis and inflammation-associated fibrosis78,80. Most notably, EMT is often coupled to the downregulation of E-cadherin, upregulation of cell–extracellular matrix (cell–ECM) adhesions, deposition of ECM proteins, and enhanced cell motility. This transition is thought to enable tumor cells to disseminate toward the lymph nodes and vasculature79. Alternatively, inflammation-associated EMT is necessary for tissue repair and ceases at the end of the inflammatory response. However, inflammation during chronic infections can lead to sustained EMT, tissue damage, and organ fibrosis81,82.
Chlamydia infection can promote EMT in different epithelial subtypes and through multiple pathways. Oviduct epithelial cells infected with C. trachomatis serovar D increase the expression of microRNAs (miRNAs) that promote EMT83. By immunofluorescence microscopy, infection downregulates the expression of E-cadherin while upregulating the EMT markers smooth muscle actin (SMA), the matrix-degrading enzyme matrix metalloproteinase-9 (MMP9), fibronectin, and the transcription factors SNAIL1/2 and ZEB183,84. Similar results were observed in conjunctival epithelial cells infected with C. trachomatis serovar B. Infection increases TGF-β expression which signals through SNAIL/ZEB2 and drives the downregulation of E-cadherin and upregulation of fibronectin and SMA. These changes may also involve epigenetic modifications as methylation of E-cadherin, fibronectin, and SMA genes were observed85.
More recently, a global phosphoproteomic and transcriptomic analysis in ectocervical epithelial explants infected with C. trachomatis serovar L2 indicated that infection activates cell signaling pathways to promote an EMT-like signature43. The infection initiates at the top of the stratified epithelia and progresses toward the basal layer, suggesting that altering cell–cell junctions during EMT may provide access for subsequent rounds of infection. Indeed, downstream of the MAPK pathway, the transcriptional factors ETS1 and ERF promote EMT in infected cells, leading to the downregulation of E-cadherin and increased cell motility and invasion43. Collectively, these observations suggest that Chlamydia can promote the transformation of epithelial cells, which may contribute to cancers of the cervix and uterus. However, these results must be confirmed in vivo and in the proper tissue context.
Extending these observations to a robust animal model of infection has remained challenging. Although the mouse-adapted C. muridarum is used extensively to study the host immune response in mice, this Chlamydia species is less genetically tractable than C. trachomatis. On the other hand, intravaginal inoculations of C57BL/6 mice with C. trachomatis often fail to induce significant pathology because the bacteria do not efficiently ascend to the upper genital tract86. However, transcervical inoculations87 that bypass the vaginal vault and the use of more permissive mouse strains (for example, C3H/HeJ) have improved the ability to monitor the infections in the mouse upper genital tract and ensuing pathology and infertility88. C. trachomatis serovar L2 ascends to the upper genital tract in C3H/HeJ mice when inoculated intravaginally, stimulating a robust immune response and altering epithelial cell height89. Future studies combining these new infection models and techniques with both host and bacterial genetics will promote better molecular dissection of Chlamydia pathogenesis in live animals.
The recent advances in the Chlamydia experimental tool kit have brought about a new era in Chlamydia research. With the ability to specifically disrupt genes and express genes in trans, new mechanisms by which Chlamydia subverts its host will be identified. Applying these molecular tools in model systems that better mimic the in vivo physiology will significantly accelerate our understanding of the infection process, especially as these tools are applied to other C. trachomatis serovars with distinct tissue tropisms. For example, using more sophisticated infection models and defined C. trachomatis mutants, we now test how specific virulence factors promote infection and affect epithelial cell growth, function, organization, and potentially transformation. However, more work is necessary to decode the genetics of the urogenital and ocular biovars and generate the relevant in vivo models of infection that best recapitulate the host responses observed in humans. Collectively, these new tools and models can be prioritized for the identification of new therapeutic targets or harnessed for the rational design of vaccines for this clinically important pathogen.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)