Keywords
hypertension, blood pressure, MTHFR, riboflavin, clinical trial
hypertension, blood pressure, MTHFR, riboflavin, clinical trial
AE: adverse events; BMI: body mass index; BP: blood pressure; CRF: case report forms; CTSO: clinical trial support office; DAG: data access group; DM: data manager; EGRAC: erythrocyte gluthathione reductase activation coefficient; US FNB: United States Food and Nutrition Board; FAD: flavin-adenine dinucleotide; GCP: Good Clinical Practice; HIGH: Hepcidin and Iron in Global Health; MRCG at LSHTM: Medical Research Council Unit The Gambia at London School of Hygiene and Tropical Medicine; MTHF: methylene tetrahydrofolate; MTHFR: methylene tetrahydrofolate reductase; QC: quality check; PI: principal investigator; REDCap: Research Electronic Data Capture; RNI: Recommended Nutrient Intake; SAE: severe adverse events; SEM: structural equation modelling; SSA: sub-Saharan Africa.
Hypertension is a global public health problem affecting 1 billion people worldwide and up to 46% of adults in sub-Saharan Africa (SSA)1. Despite a generally very low body mass index (BMI) in the rural Kiang West district of The Gambia, the prevalence of high blood pressure (BP) (>140/90) has recently been estimated as 18.3% in adults over 18 years2. Hypertension arises from a combination of multiple lifestyle and genetic factors (many of which probably remain unknown). Its treatment requires an individualized combination of drug therapy and lifestyle changes3,4. The treatment of hypertension is generally lifelong and exerts a heavy economic burden on individuals and their families as well as on national economies. The identification of safe cost-effective treatment interventions without adverse side effects would be hugely advantageous, particularly in low-income settings with high prevalence of hypertension such as SSA.
Emerging evidence from clinical and genome-wide association studies (GWAS) links a functional polymorphism in the methylenetetrahydrofolate reductase (MTHFR) gene (rs1801133), encoding the folate-metabolising enzyme MTHFR, with blood pressure (BP) and hypertension in adults5,6. This enzyme catalyses the conversion of 5,10-methylenetetrahydrofolate (MTHF) to 5-MTHF. Variation at rs1801133 is relatively common and has 3 genotypes; homozygous “normal” wild-type CC, heterozygous CT and homozygous “variant” TT genotypes. Of these genotypes, the homozygous variant TT is strongly associated with a higher BP. The precise mechanism by which MTHFR C677T is associated with BP remains unclear.
Vitamin B2 (riboflavin) is converted to flavin-adenine dinucleotide (FAD) which acts as the essential co-factor for the MTHFR enzyme. Molecular studies demonstrate that the decreased activity of the variant enzyme is caused by reduced affinity for the redox cofactor FAD7; hence its original name as the ‘heat-labile’ variant. Work at Ulster University has demonstrated, in three separate randomised controlled trials to date, that BP is highly responsive to intervention with riboflavin and that this response is dependent on MTHFR genotype8–10. In all of these trials, riboflavin supplementation resulted in significant reductions in both systolic and diastolic BP in adults with the homozygous variant TT genotype, while at baseline (pre-intervention) we observed intermediate BP values in those with the heterozygous CT genotype compared to adults with CC (lower) or TT (higher) genotypes.
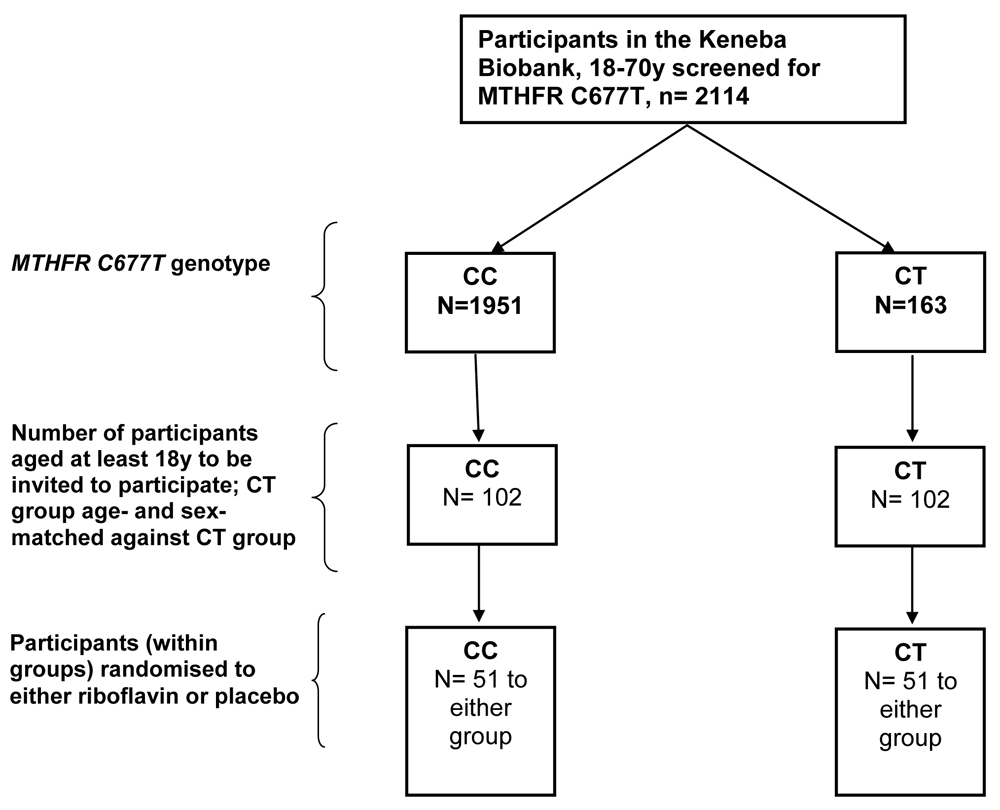
In the Keneba biobank11, a total of 2114 individuals between the ages of 18 and 70 were genotyped for rs1801133 of whom 1951 (92.3%) and 163 (7.7%) had the CC and CT genotypes respectively, while none had the TT genotype. We hypothesize that the almost universal severe riboflavin deficiency seen in most Kiang West residents of The Gambia (assessed historically12) and more recently13 will a) mimic the effect of the TT variant and b) exacerbate the effect of the CT genotype based upon the following argument. In vitro studies show that the TT variant is only 30% saturated with FAD compared to CC and the CT is 84% saturated14. Riboflavin deficiency is a major problem in Kiang West district with typical values for erythrocyte gluthathione reductase activation coefficient (EGRAC; gold-standard riboflavin biomarker) of >2.0 (compared with indicative values of <1.2, 1.2-1.4 and >1.4, defining normal, suboptimal and deficient status, respectively), indicating that EGR is only 50% saturated by FAD. By analogy, we expect these levels of severe deficiency to lower levels of MTHFR activity by ~50% in CC the genotype and a further 16% in CT variants. This is the basis for the anticipated efficacy of riboflavin supplementation.
The adult recommended nutrient intake (RNI) for riboflavin is 1.3 mg/day for men and 1.1 mg/day for women15. There is no known toxicity (due to a renal threshold whereby unused riboflavin is excreted rapidly in the urine) and the US FNB has noted that there have been no known adverse effects, even at intakes of 400 mg/day for at least 3 months16. We propose to use just 5 mg/day to replenish body stores and attempt to saturate the CT genotype enzyme as well as aligning the dose with previous trials.
The possibility that riboflavin deficiency represents a new, easily-correctible causal factor in hypertension in SSA would require further large-scale interventions if this study yields encouraging results.
The primary objective is to compare the effect of 16-weeks of riboflavin supplementation (5 mg/day) versus placebo on systolic and diastolic BP.
The secondary objectives are:
a) to test for possible effect modification in CC vs CT genotypes for the MTHFR C677T polymorphism on BP response to intervention
b) to describe the cross-sectional associations at baseline between BP (continuous variable and proportion with hypertension, >140/90 mmHg) and riboflavin status (assessed by the EGRAC functional biomarker test) and interaction with MTHFR genotype
The primary outcome is change in BP at 8 and 16 weeks compared to baseline in response to riboflavin supplementation versus placebo. We will also determine the response of the riboflavin biomarker to intervention, determined (before and after riboflavin supplementation) using the EGRAC method, considered to be the gold standard method to assess biomarker status of riboflavin.
This is a phase 2 recall-by-genotype, randomised single-blind placebo-controlled micronutrient supplementation trial. We will invite all individuals between the ages of 18 and 70 in the Keneba biobank with the CT genotype and a similar number of age- and sex-matched CC homozygotes to participate in the study (Figure 1).
The study protocol was reviewed by the Scientific Coordinating Committee of the MRCG at LSHTM and has received ethics approval from the Joint Gambia Government/MRC Ethics Committee (SCC1518). Approval was also obtained from the National Medicine Regulatory Authority of The Gambia.
Participants must meet all of the inclusion criteria and none of the exclusion criteria to be eligible to participate in the trial.
Inclusion criteria
Aged between 18–70 years
Has provided appropriate ethical consent for involvement in studies relating to genetics.
Has available genotype data in the Keneba biobank needed for the current study
Available for the duration of the intervention period
Exclusion criteria
Sensitization of community elders, youth and women groups and respective village residents within the Kiang West district was conducted by the study team prior to recruitment of study participants. During community sensitization, community elders of respective villages were asked to identify literate people to act as impartial witnesses during the consenting process of illiterate potential participants.
Individual consent for the study will sought by trained field assistants after explaining the project in full detail, covering all aspects of the study as laid out in the approved study information sheet. Literate participants will be given a copy of the information sheet while illiterate participants will have the full information sheet read to them in the language they understand in the presence of an impartial witness. The fieldworkers answer any questions that arise, and participants are also be given the possibility to obtain further clarifications and explanations by speaking to one of the study investigators if they wish. Participation is entirely voluntary and individuals who are not able to give consent are not enrolled. If participants agree to participate, written informed consent is then obtained. Assessment of informed consent understanding is performed using a specially designed form of 10 questions derived from the participant information sheet (available as Extended data17).
The Keneba biobank is used to identify potential participants. After the exclusion criteria are applied and non-consenters are taken into account, we aim to enrol at least 102 participants (see sample size calculations below) with the CT genotype and a similar number of age and sex-matched participants with the CC genotype. A randomisation code, written in Stata, would select each of the participants with the CT genotype and match them to participants with the CC genotype as randomly and equally as possible. This will ensure that all participants with the CT genotype would be age and sex-matched to several possible individuals with the CC genotype. Since each consented CT genotype participant has several possible matches with the CC genotype, consent would be sought until it is obtained from any of the possible matches. Thus if the first participant approached with the CC genotype consents to participate, all other possible matched participants would not be sought after for their consent.
Following consent, the participants’ Biobank IDs would be linked to their Study IDs and allocated to receive either riboflavin or placebo by block randomization (of varying block sizes). To ensure balance between the two genotypes, we shall randomly allocate 50% of those with the CT genotype to receive the riboflavin supplement. A programme will perform the randomisation in a 1:1 ratio and produce randomization envelopes to be used. This randomisation is such that for each CT genotype participant that receives the riboflavin supplement the matched participant with the CC genotype is automatically allocated the placebo and vice versa. The randomisation codes and database are accessible only to an independent statistician and data manager.
Field nurses and field assistants measuring BP and dispensing investigational products to participants, and laboratory staff will be blinded to the genotype of the study participants and to the identity of the treatment arm to which a participant is assigned from the time of randomization to the time of unblinding. Details of matching will also not be revealed to them. The placebos are designed to be indistinguishable from those containing active drugs. The products will be pre-packed with the participant’s name and study ID on a weekly basis by an unblinded study nurse and clinical trial assistant following a study specific protocol and handed over to field assistants who will distribute them to study participants. Double-blinding is not possible because subjects randomized to the riboflavin will have yellow urine, a harmless outcome that will be explained to them at the start of the study and told not to discuss this with any member of the study and other MRCG at LSHTM staff.
In this trial, we will ensure that sealed envelopes containing the individual treatment allocation are kept at the Medical Research Council Unit The Gambia at London School of Hygiene and Tropical Medicine (MRCG at LSHTM) field station in Keneba in case of the need for emergency unblinding. The code would be broken in cases of severe adverse events for which the product needs to be known for individual participants.
The blinding code will be kept by the independent statistician and will only be broken after the database lock.
At the initial visit, socio-economic data and medical history will be obtained by a field staff using a questionnaire. Anthropometric measurements (height, weight, waist and hip circumference, skin-fold thickness and body composition by bioelectrical impedance analysis) will be taken. Thereafter, BP will be measured with an automated Omron 705IT device (Omron, Kyoto, Japan) in triplicate, each in a seated position and 5 minutes apart, using a standard protocol by the same investigator who will be blinded to genotype and treatment group. The mean of the second and third measurements will be used for analysis. A 10ml blood sample will be collected for laboratory assessment of vitamin B status. During the intervention phase of the trial, we will supplement participants with 5mg/day of riboflavin or a matching placebo for 16 weeks. The pills will be supplied every 10 days with instructions to return any unused pills. Another BP measurement following similar protocol as before will be taken at 8 weeks as well as at 16 weeks post- intervention. Finally, a 10ml blood sample will be collected 16 weeks post-intervention. Blood samples will be analysed for riboflavin (by EGRAC), homocysteine, red cell folate, cobalamin and pyridoxine. Participants will be followed-up after the intervention period for a further 3 weeks to monitor AEs. Details of the study schedule are summarised in Table 1.
The investigational products to be used in this study are manufactured by Advanced Molecular Research, Calgary, Canada (https://aor.ca/) following Good Manufacturing Processes and quality control procedures. The products are in the form of capsules, are physically indistinguishable from each other and having the following excipients: microcrystalline cellulose, hypromellose (capsule shell) and chlorophyll (color of the capsule shell) with the supplements (as opposed to placebo) containing 5mg of riboflavin. 10 green capsules are packed in a clear plastic blister and stored as per manufacturer instructions at the MRCG at LSHTM Field Station in Keneba. All used blisters and unused pills will be recovered for accountability purposes as required by local regulations.
The trial will be monitored by personnel of MRCG at LSHTM Clinical Trial Support Office as per the sponsor’s regulations. An initiation visit between the monitors, Principal Investigator (PI) and study team was conducted to approve the start of the study. The purpose of this visit is to review ethical approval, the trial master file and required documents (logs, study specific procedures, data collection tools), investigational products, and ensure that study specific training and appropriate facilities are available. An interim visit will be conducted 1 month after the start of the study and then after 6 months. These visits will include the following: review of current status and recruitment rate, verify that the study is conducted in accordance with approved protocols and regulatory requirements and perform source data verification. A close out visit will be conducted at the end of the study to ensure completeness of all essential documents and archiving arrangements, and appropriate distribution of all remaining study products and supplies.
The trial will provide 5-mg riboflavin pills or a matching placebo. Riboflavin is safe and because it is water soluble, any excess will be excreted via the urine. No adverse events were observed in studies by our collaborators8–10 and the US FNB has noted that there have been no known adverse effects of consuming 400 mg/day (80 fold higher than our proposed dose) for at least 3 months16.
The only potential adverse risk identified in this study relates to blood collection, i.e. pain or discomfort, bruising and infection. The total amount of blood to be collected is 20 ml (10 ml at baseline and at the end of the study) which does not pose any health risk. Our study nurses are sufficiently trained and standard protocols will be used to avoid infection.
Participants who will receive the riboflavin supplements will have their riboflavin stores improved and possibly replenished. Other participants who were randomized to receive placebo will be offered riboflavin supplementation at the end of the study.
Format and scale of data: The database software to be used in this trial is REDCap. Trial staff trained on GCP and electronic case report forms (eCRF) will be authorized to collect data and other trial specific documentation. Training on REDCap was conducted and they will continue to be updated through in-house training. A web and mobile version of the application installed on mobile tablet devices, smart phones, laptops and PCs will be used at each stage of data collection.
The trial data manager and database developer in consultation designed the eCRFs with input from the PI/sub-investigator and research clinician according to MRCG at LSHTM’s CRF Development standard operating procedure. The eCRFs have built-in quality check (QC) process which aims to minimize the rate of errors/missing fields and enhance data quality. Other quality control measures will include running queries by the DM that will flag discrepancies in the form of missing values, records and outliers. The database developer developed all data entry screens and all reporting functionalities. The final data format can be downloaded to Excel, PDF, and common statistical packages (SPSS, SAS, Stata, R).
The eCRFs will include question fields which are either single or multiple checked, drop-down single selection answers, binary (yes/no) fields, numerical fields and free text. REDCap branching tools will be utilised to guide data collection making sure data is collected on the required activity at each visit. For instance, sample collection and processing data should be allowed to be collected only at visits 00 and 11. Study time points after recruitment and randomisation are categorically numbered 0–14 (Table 1). Call lists will be generated to help organize study visits. Strict validation and verification rules will be adhered to when completing the eCRFs. All data collected on mobile tablets collected offline will be synced directly on MRCG at LSHTM dedicated servers accessible only by users within a given Data Access Group (DAG) managed by the software. User access groups for the trial will be granted to trial staff based on individual responsibilities and privileges.
System validation: All validation, range logical checks will be applied as required to maintain data integrity. Demographic and clinical data will be imported into the study database but all personally identifiable fields will be marked in REDCap to prevent them from being exported as part of datasets. Data export options from the clinical database include CDISC ODM XML, a vendor neutral, platform independent format for interchange and archive of data collected in clinical trials. This should ensure sharing and long-term validity of data.
All data collection instruments will be continuously reviewed at the various stages of development to ensure that data collected meet the required standards in line with study analysis plan. Data entry screens are designed with rigid validation and logical checks, skip patterns and validations to accurately guide data collection. Study specific procedures (SSPs) and the protocol will guide data collection at the field and in the lab.
Instructions will be embedded on the eCRFs. As part of data supervision, status of the record’s data form completeness, denoted with a colour; red incomplete; yellow unverified; green complete will allow data to be firstly submitted in complete status. The supervisor ensures these records are verified and record status changed to verified in a timely manner.
Data validation: Validation checks will be enforced real-time at each stage of data collection which will be programmatically built into the database application to ensure accuracy, consistency and completeness. Primary data will be generated from the field and lab which shall be verified by the study PI or delegate and supervisors.
Other QC checks will be routinely conducted to ensure data collected is of high quality. Participants and visits will be programmed using REDCap calendar to ensure that they are seen on scheduled time points.
Mandatory fields must be completed or flagged for data entry to proceed. Data fields are mainly univariate, allowing only one acceptable value or option. Multivariate checks will be implemented where required. Date fields will be formatted as dd-mm-yyyy and pre-determined ranges applied. Every field will be formatted with the right input masks (e.g. visit number can be masked as regexp: /\d {2}/, to allow two-digits like 01).
Data coding: An up-to-date data dictionary will document the study metadata. The data dictionary specifies the database structure normalised to an appropriate level. It specifies database tables, variable names, description types an all properties including all data coding. Categorical and float data will be coded using standard conventions (e.g. 1 = Yes; 0 = No) and range checks on numerical data will ensure plausible data is collected in line with the analysis plan. XML features in REDCap keeps a downloadable up-to-date study metadata.
Data capture: For audit purposes, all new and modified records will be logged with the username of the person collecting the data. Data will be captured at each stage using REDCap mobile application installed on tablets that allow data to be collected offline and synced later through a Wi-Fi or mobile data connections. Appropriate rights are allocated and managed by the DM to users. The ability to view any data will be limited and permissions granted by the data manager through DAGs.
Query resolution: Due to the real time nature of data collection, queries/errors are expected to be easily detected and resolved immediately. It will be mandatory on all study staff to raise and attend to queries in a timely fashion. In addition, the monitors may raise queries with the team using the same process. A log of all queries handled by the data manager or delegate at any point can show how many queries are raised, resolved, or pending.
Data transfer and consolidation: Extracted data requested by the study team will be in the form of CDISC ODM XML or Excel. This will be made available to the recipient through the appropriate medium or as requested by PI. It will be carried out as and when required. Data to be transferred to study PIs would be zipped and password-protected. All data transfers will conform to MRCG at LSHTM data transfer procedures.
Safety data reconciliation: Serious adverse events (SAEs) are least expected in this trial but they will be assessed by research clinician and immediately logged and reported to the Clinical Trials Support Office (CTSO) by email alerts. Standard questionnaires will used to report and follow-up SAEs.
Protocol deviations will be reported and kept in the Trial Master File.
Database security: All persons dealing with personal data are responsible for ensuring the security and safety of these data. Strict access controls will apply both to the servers and the database. Access to the database is controlled by the Data Manager and any requests for access would be documented.
Study closure: Lost to follow-ups would be assessed and after the final trial monitoring visit is completed, the following targets are expected to be met:
That all data is entered into the study REDCap database and all records marked as complete
All AEs reported and SAEs followed
All queries are resolved and data cleaning completed
Source document verified
Access rights revoked
The database would be locked in due course and all accesses denied.
Sample size rationale:This pilot trial is constrained by the number of available eligible and consenting adults carrying the MTHFR 677 CT variant. We estimate this will be 102 to be matched to an equal number of CC controls. Using standard deviation estimates from our own and published literature (14 mm for systolic BP and 10 mm for diastolic BP) we will have 95% confidence (at p<0.05) of detecting differences of 3.9 mmHg for systolic BP and 2.8 mmHg for diastolic BP.
Statistical analysis plan:A preliminary evaluation of data will be conducted to identify issues that will affect our approach to the primary analysis. This will specifically include the following:
summarize the amount of missing data for key variables,
summarize time between baseline and follow-up key measurements
examine the distributional characteristics of key variables in order to detect potential data entry/reduction errors
We will also carry out tabulation of descriptive statistics for key variables (including missing observations) and histograms, quantile plots of baseline levels and follow-up measures and changes in those measures will be assessed.
We will also describe the study process to assess whether there is a difference between the trial results compared to the pre-trial plans. “Table 1” describing the baseline characteristics of the riboflavin and placebo arm of the trial as well as a CONSORT diagram will also be constructed.
The statistical approached to be used to evaluate the primary and secondary study end points are discussed below:
We shall use regression to compare differences in BP at week 16 between the two arms of the trial after adjusting for the BP at week 0. The models are summarized below
for i = (1 for riboflavin and 0 for placebo)We would perform diagnostics to investigate normality and heretoskedasticity so as to perform a transformation as necessary.
The analyses on BP would be for systolic and diastolic separately. We shall use structural equation modelling (SEM) to predict a latent variable that could reduce the dimension from two to one variable that could reasonably summarize overall change in BP. This would be done by predicting a latent variable for both systolic and diastolic BP at week 0 and week 16 before comparing the two arms using regression as summarized in the model below.
Assessing the relationship between the systolic and diastolic BP with the overall BP would be assessed to see if the relationship is clinically plausible. If it is, then it can be used to test whether, holistically, BP at week 16 is different between the riboflavin arm and the placebo arm, adjusting for overall BP at week 0.
Missing data: Missing data less than 5% (below the 102 participants required for adequate power in the sample size calculation) would be considered acceptable to do a complete case analysis. In the event that the missing data is or above 5% we shall use Multiple Imputation to minimize bias.
Interpretation: We shall reject the null hypothesis that there is no difference between the riboflavin arm and the placebo arm at a 5% significance level. A statistically significant result between the arms where the mean BP (separated or overall) for participants that received riboflavin is lower than the mean of the participants that received placebo would mean that riboflavin successfully lowers BP 16 weeks after administration.
We shall use regression to ascertain whether there is effect modification of the intervention due to the genotype. The models are summarized below
for i = (1 for riboflavin and 0 for placebo)Similarly diagnostics would be performed to assess none of the assumptions of regression were violated and ensure that proper methods would be used to address any violations.
Interpretation: We shall reject the null hypothesis that there is no effect modification at a 5% significance level.
We shall use regression to ascertain whether there is an association between EGRAC based riboflavin status and systolic and diastolic BP at both week 0 and week 16. This is when we are considering the BPs as continuous. The models are summarized below.
The models that use a BP cut-off of 140 mmHg for systolic and 90 mmHg for diastolic are similar to the ones above.
for i = (1 for 140 mmHg and above and 0 for less than 140 mmHg) j = (1 for 90 mmHg and above and 0 for less than 90 mmHg)Diagnostics would be performed to assess and correct violation of any regression assumptions as appropriate.
We shall also assess the effect modification by the MTHFR genotype using both continuous and dichotomized BP measurements. These models would also be diagnosed and violations addressed accordingly.
Interpretation: We shall reject the null hypothesis that there is no effect modification at a 5% significance level.
High BP is a public health problem, especially in SSA. The causes are not fully understood in the vast majority of cases and commonly result from a combination of lifestyle and genetic factors. The treatment of high BP is often very expensive and sometimes not affordable especially in poor communities. The development of low-cost, effective treatments would therefore be hugely beneficial.
It is possible that the severe riboflavin deficiency observed in the Kiang West district and possibly in other parts of SSA may be contributing to the rising prevalence of high BP, especially among those with a genetic predisposition for developing hypertension owing to MTHFR 677CT genotype. The study will represent the first step in helping us to understand if riboflavin supplementation helps to control blood pressure in this population group. If improvements in BP were achieved in this study and proven in large-scale intervention, it would offer a novel means of controlling hypertension that would be safer, more accessible and more cost-effective than current treatment options.
A total of 122 participants have been randomised into the study at the time of submission.
Figshare: RiboBP_SPIRIT Checklist.docx. https://doi.org/10.6084/m9.figshare.12627902.v117.
File ‘Prentice_SCC 1518_ICD_version4.0.docx’ contains the participant information sheet used in this study.
Figshare: SPIRIT checklist for ‘Effect of riboflavin supplementation on blood pressure and possible effect modification by the MTHFR C677T polymorphism: a randomised trial in rural Gambia. https://doi.org/10.6084/m9.figshare.12627902.v117.
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)