Keywords
SNP, Disease, Python, Bioinformatics
SNP, Disease, Python, Bioinformatics
With the extending and deciphering of genomic data produced by sequencing technologies and the Human Genome Project1, much information has been discovered, such as exons, introns, domains, coding sequence, and non-coding sequences. Mutated signals have attracted tremendous attention because of their crucial impact in altering gene expression, in particular, single nucleotide polymorphisms (SNPs)2, which also act as gene molecular-markers for an associated trait. Additionally, the existence of a specific SNP can indicate precisely what disease is foreseen and the possible drugs to treat it, which is considered the ultimate goal of pharmacogenomics.
Pharmacogenetics, or its inclusive version pharmacogenomics (since it covers proteomic, genomic, epigenomic and transcriptomic effects on disease and drug response), has produced a vast body of research since 19973, due to its key importance in personalized medicine through investigating how far genetic variations (e.g. SNPs) are involved in disease development and determining drug targets. Thereby the safety and efficacy of an individualized drug therapy can be improved. Since the relationships between molecular data pertaining to patients and their disease phenotype are complex and difficult to determine manually, scientists have begun to develop and enrich the bioinformatics knowledge base with more sophisticated and accurate molecular tools to detect genetic variations for example, SNPector is a recent tool developed by the authors to detect SNP effect in drug response and disease development4. This will allow interpretation of how these tiny variations may cause direct errors, e.g. X-SNP is a common SNP type that gives rise to premature termination codons that halt gene expression5. Some SNP types involved in alteration of the protein production process cause disease6, while other regulatory SNPs7 may disrupt pathways causing cascade errors that lead to collapse of a whole pathway thereby causing disease development.
Elucidations about SNPs play a central role in providing recommendations for practicing physicians. In addition, a wide range of research fields have arisen out of pharmacogenomics, such as vaccinomics, which study aberrant immune responses to vaccines based on genetic makeup. For example, a specific SNP in the TLR3 gene was found to be responsible for the reduction of humoral immune responses and cell-mediated immunity to the measles vaccine8. Nutrigenomics is the science of gene–nutrient interactions, which involves research methods and clinical implementation to detect and treat nutrient-related diseases. One of the best known examples of nutrigenomics is lactase persistence, in which the gene encoding lactase is expressed past weaning. Lactase persistence in Europeans is caused by a polymorphism called “C-13910T” in the lactase phlorizin hydrolase gene promoter9, and the lack of C-1390T in many adults can lead to severe gastrointestinal discomfort and diarrhea resulting from ingesting milk due to the inability to metabolize lactose10.
Here we introduce Pharmosome, a web-based, user-friendly and collective database for more than 30,000 human disease-related SNPs, with dynamic pipelines to explore SNPs associated with disease development, drug response and the pathways shared between different genes related to these SNPs. Pharmosome implements several tools to design primers to detect SNPs in large genomes and facilitates analysis of different SNPs to determine relationships between them by aligning sequences, constructing phylogenetic trees, and providing consensus sequences illustrating the connections between SNPs.
We collected SNP-related data (e.g. SNP ID, annotation, pathway and phenotypes) from PharmGKB11, NCBI12,13, Ensemble14, DiseaseEnhancer15, GeneCards16 and Reactome17 in tab-delimited format in order to link between different available information. The collected data were categorized into four main sub-databases: SNP, Gene, Chemical and Disease. The Python 3+ programming language was utilized to read, select, filter and sort the data and to link the Python scripts with HTML and JavaScript codes.
Data collection. About 50% of the data was downloaded from PharmGKB, which is considered the most common database for SNP annotation. The data comprises the associated phenotype, the clinical perspectives and, considering storage space limitations, the remaining data are imported on-demand to use later by a set of Python functions we built to get access to the Application Programming Interface (API) of different databases. These other databases include GeneCards, DiseaseEnhancer, Ensembl and Reactome; thereby a user can return specific data using preset IDs and then export this information to the HTML interface. The number of data entries collected by Pharmosome is shown in Table 1.
Data fetching and exporting. We constructed a Python module containing 12 functions. These functions connect in different ways to import the data from the databases above either from tab-delimited files or using APIs. User-requested information is extracted and exported to the Pharmosome web interface. The Django web framework was used to build the Pharmosome web interface with HTML programming language. We used Django to build functions that receive the user input from the Pharmosome interface, process requests using a Python script, and finely export the result to the interface.
return render(request, 'WebInterface.html', context={})
Google Chrome browser is recommended to use the Pharmosome web interface (https://pharmosome.herokuapp.com/), but other internet browsers can also be used.
SNP sub-database. In the SNP sub-database, users can enter the ID of a SNP (e.g. rs141033578) and receive output data about its related gene, chromosomal location, gene bands, summary of the normal function of the gene, the gene part responsible for enhancing the disease occurrence, pathway of defective gene (if available), gene transcripts and different splicing variants. Pharmosome also provides information that can be used to retrieve data of recent studies (specifically, SNP reference nucleotide and alternative nucleotide data and an explanation of how the SNP contributes to disease development and drug response). For example, an SNP in GSTP1 was associated with overall survival in 107 patients with metastatic colorectal cancer who received 5-FU/oxaliplatin combination chemotherapy that caused the replacement of isoleucine with valine at amino acid position 105 of the protein, which is known to substantially diminish enzyme activity18.
Disease sub-database. The Disease sub-database allows users to search for disease data collected from KEGG Disease and PharmGKB databases by entering the disease name. The search result is a list of genes responsible for the disease, the ID of the SNP occurring in this gene, a description of clinical annotations related to this gene and the gene-specific chemical used in the treatment. The sub-database can be used, for example, to identify the SNP, gene and chemical related to coronary artery disease caused by a high frequency of a particular polymorphism in the PLA2 gene. This gene encodes glycoprotein IIIa, which is associated with a high prevalence of premature myocardial infarction19,20
Chemical sub-database. The Chemical sub-database has data from four sources: KEGG, PharmGKB, DrugBank and ChemSpider. After users enter a chemical name, the output is a list of the chemical name, trend and generic names, structure, description and pharmacodynamics. For example, by inputting bortezomib (a proteasome inhibitor), users receive output data relating to the clinical success of bortezomib, which established the ubiquitin (Ub)+proteasome system as a key therapeutic target in multiple myeloma21,22.
Gene sub-database. The Gene sub-database links between different sources (NCBI GenBank, Ensemble, Reactome and DiseaseEnhancer). In particular, the DiseaseEnhancer dataset represents a new approach that determines the gene part that is responsible for enhancing occurrence of disease. NCBI provides information about gene name, location, a summary of gene function and chromosomal location. Reactome displays the pathway in which genes are involved and gives an overview description. The Gene sub-database can be used to identify the pathway, splicing variants, disease enhancing region, genomic and proteomic expression profile or even to get general information. An example of this would be looking at the SLCO1B1 gene, for which various groups tested the hypothesis of whether polymorphisms in SLCO1B1 affect pharmacokinetics and the effects of drugs in humans23.
SNP collector. The SNP collector is a tool within Pharmosome that is designed to find all SNPs present on the gene, related to disease, associated with the chemical compound. Users can choose between different options to collect SNPs and their clinical annotations and chemicals related to each SNP. This tool can be used to find SNPs or other information. For example, it could be used to detect the bond work which occurs on location 118 of the mu opioid gene, which was 3 fold more vigorous than the wildtype in its interaction with b-endorphin Other regulatory-SNPs in this region of the gene can be linked to other phenotypes24.
Pick primer. The detection of SNP existence in a DNA sample taken from patients depends on designing an appropriate primer. Primers should be compatible with the flanking sequences of SNP. As the presence of an SNP in the genome may result in disease and affect the choice of drug, there is a need to detect the presence of SNP e.g. for early disease diagnosis. The pick primer tool within Pharmosome has the important function of designing primers to detect an SNP in the genome by retrieving the SNP sequence record from the NCBI database, locating the SNP position and designing primers 50 bases before, after and within the SNP sequence.
SNP phylogeny. As discussed in previous sections, there is always some relationship between different SNPs due to the complicated interaction network between different genes. In order to determine how these SNPs are related to each other, the SNP phylogeny tool constructs a phylogenetic tree that illustrates the relationships between different SNPs25 by downloading SNP and flanking sequences and commencing multiple sequence alignment to determine how far each sequence is related to others. This function could be used clarify connections in studies, such as that of Thompson et al. that showed an association of 43 SNPs in 16 genes with the response drug of atorvastatin26.
Figure 1 shows the flow of information to meet the needs of users.

Pharmosome deploys seven sub-databases and tools. Our approach during the building of Pharmosome, is to achieve the easiest usage. We designed each tool and sub-database to receive the user input with minimum required parameters (as shown in Table 2). Users enter the target input they require and the Pharmosome interface will automatically redirect to another page that shows the user the output results. Figure 2–Figure 5 show output on the Pharmosome web interface.
| Function | Input required | Input example | Output description |
|---|---|---|---|
| SNP sub-database | SNP ID | rs75527207 | SNP related disease and drug response annotation |
| Disease sub-database | Disease Name | Heart Failure | Gene involved in disease, chemical used in treatment, and SNP causing the disease (Figure 1) |
| Chemical sub-database | Chemical Name | Ivacaftor | Description of target disease (Figure 2) |
| Gene sub-database | Gene Symbol | CFTR | Gene annotation (Figure 3) |
| SNP collector | Gene, Chromosome, Disease, or Chemical | CFTR, 11, Heart Failure, or Ivacaftor | List of SNPs (Figure 4) |
| Pick primer | SNP ID | rs75527207 | Forward and reverse primer |
| SNP phylogeny | SNP List | rs113993960 rs2853741 rs1045642 rs3857532 rs10817464 rs16969968 | Illustration shows the phylogenetic tree of the input SNPs |
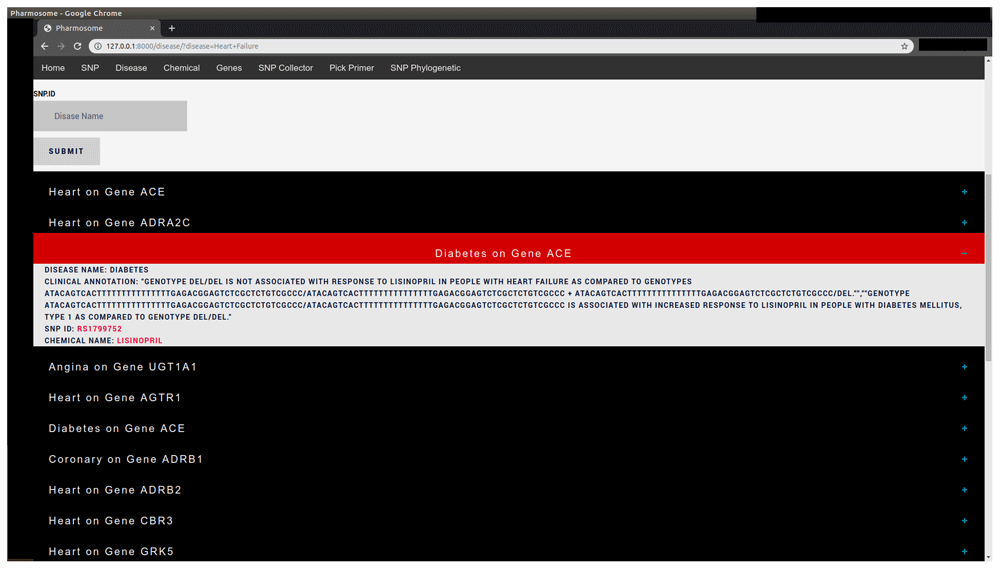
The results consist of list of drop-down menus. Each menu describes disease annotation and the associated gene to the disease.
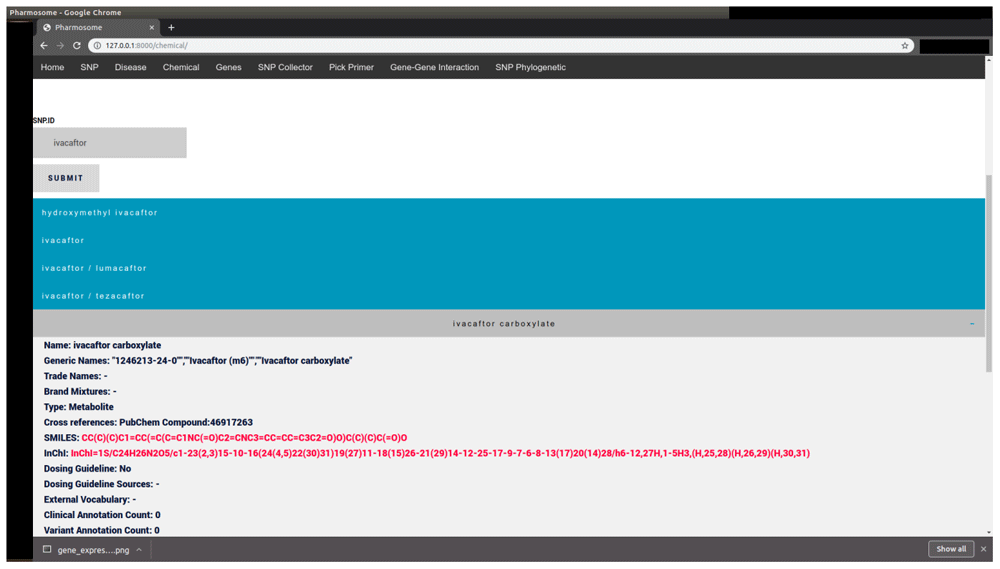
The results consists of a list of drop-down menus. Each menu describes Drug/Chemical annotations.
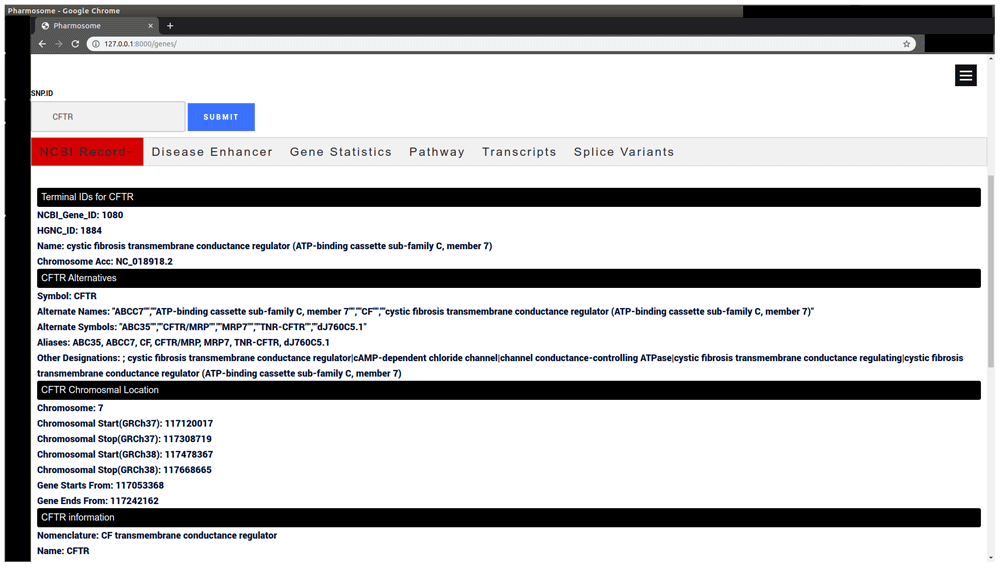
The results consist of a navigation bar, and each button expands to a different annotation.

The results consists of a list of SNPs located on this gene.
In this study, we introduce Pharmosome, an integrative and collective database for exploring and analysing human SNPs and the associated disease and drug response. Our tool deploys various functions to determine the relationships between different SNPs, construct the consensus sequence between different SNPs and to determine the pathways shared between different genes. Pharmosome also includes sub-databases to simplify, link and display data about gene functions, pathways, transcriptomes of genes, different splicing variants, clinical annotation, chemical structures and annotations of chemicals involved in the disease. The returned data are informative, user-friendly and easy to navigate. Pharmosome was written in Python 3.5, HTML and CSS with the implementation of Django (Python library) to design links between Python scripts and other languages.
Pharmosome web interface: https://pharmosome.herokuapp.com/
Source code available from: https://github.com/peterhabib/Pharmosome_Web
Archived source code as at time of publication: http://doi.org/10.5281/zenodo.358319127.
License: MIT
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)