Keywords
Progressive Familial Intra Hepatic Cholestasis Type-2, CRISPR-Cas9,AAVS1 site, human ABCB11/BSEP, MsbA, E. Coli, Endotoxin, Lipid A transporter, Pribnow-Schaller Box, cloning
Progressive Familial Intra Hepatic Cholestasis Type-2, CRISPR-Cas9,AAVS1 site, human ABCB11/BSEP, MsbA, E. Coli, Endotoxin, Lipid A transporter, Pribnow-Schaller Box, cloning
Progressive familial intrahepatic cholestasis type-2 (PFIC2), a severe liver disease which is familial, neonatal, progressive and often fatal which results from a mutation of ATP binding cassette subfamily B member 11 (ABCB11) gene which codes for an ABC transporter bile salt export pump (BSEP)1,2. Mutations in ABCB11 gene results in accumulation of cytotoxic taurocholate and other cholate conjugates leading to progressive hepatocyte destruction1. Currently the definitive cure for PFIC2 is liver transplantation, which is limited by suitable donor organs. Gene therapy, allogenic hepatocyte transplantation3 and autologous transplantation of hepatocytes/liver organoids differentiated from ‘gene corrected’ induced pluripotent cells could be future options4,5. Adeno-associated virus (AAV) is so ubiquitous in man and animals that about 30% of the world population are positive for this virus and to date no disease is proven to be associated with this virus6–9. In our study we inserted ABCB11 gene at the AAVS1 site using CRISPR-Cas9 tool (Figure 1) in HEK293T cells and a fibroblast line.
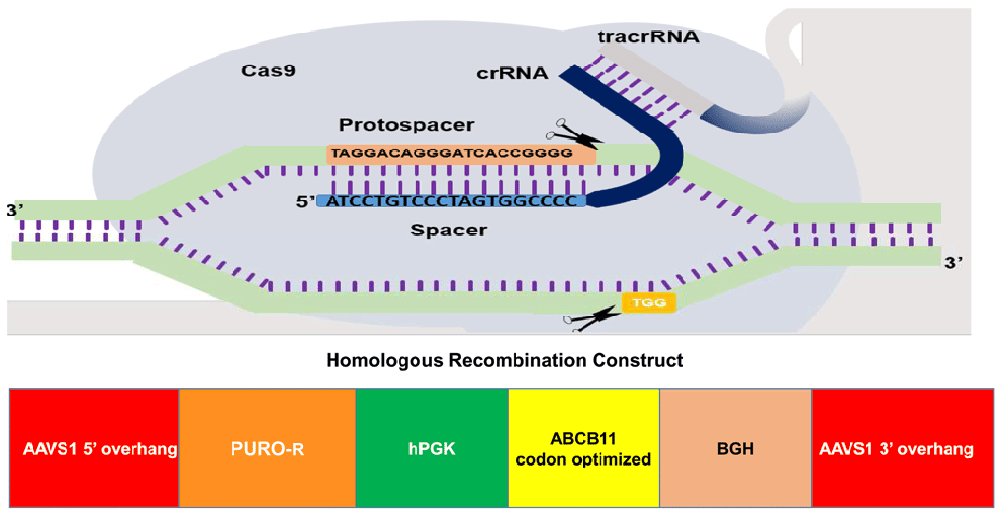
The gene of interest (ABCB11) was flanked by 5’ and 3’ overhangs which are homologous to the AAVS1 site in chromosome 19. ABCB11 gene was driven by human promoter phospho-glycerol kinase.
We PCR-amplified the 3966 bp ABCB11 (from cDNA prepared from total RNA of human liver tissue) using multiple overlapping primers (Extended data, Supp. Table 1)10 which were assembled by overlap extension PCR11. Phusion DNA polymerase (NEB, US Cat. #M0530L) was used as per the manufacturers protocol. Annealing temperate for all PCRs unless otherwise stated was 60°C C for 20s and an extension time 30s/kb at 72°C. Initial denaturation was carried out at 98°C for 30s and 5s in subsequent steps. In general, we used 24 cycles to amplify PCR products for cloning (and 32 cycles for other PCRs). We used 1.0 unit of the enzyme per 50 µl PCR reaction. The product was cloned in ‘donor’ vector having AAVS1 recombination overhangs on both ends and ampicillin resistance for selection (Extended data, Supp. Figure 1)10. The upstream (5’) overhang sequence (803 bp) in the vector was homologous to a sequence (NCBI Sequence ID: NC_000019.10, 55115768 to 55116570) inside the PPP1R12C gene (Figure 1), which would be in-frame with a 2A ribosome skip sequence and puromycin resistance gene if insertion of the construct happens by homologous recombination with the target site. This is region was followed by a poly adenylation signal and the downstream (3’) overhang sequence which was the continuation of the 5’ overhang (837 bp). We inserted the ABCB11 sequence driven by human phospho-glycerol kinase (hPGK) promoter between these overhang sequences. DH5α cells, JM 109 and One Shot™ Stbl3™ Chemically Competent E. coli cells (ThermoFisher Catalog#:C737303) were used for transformation and cloning.
Bacterial promoter prediction was done using BPROM, a prediction tool for bacterial promoters12. DNA or protein sequence comparisons were done using the appropriate tool from NCBI BLAST platform13. Primers were designed manually or using NCBI Primer Blast14.
We designed two guide RNAs targeting the AAVS1 site (Figure 1) and cloned in two vectors expressing SPCas9 and SACas9 at BsaI sites (Extended data, Supp. Figure 2)10 following the standard gRNA design, cloning protocols and resources15. Off-target analysis was done using the tool Custom Alt-R® CRISPR-Cas9 guide RNA16.
HEK293T, HepG2 and FS1 (fibroblast) cells were grown in high glucose DMEM (Hi-Media Lab, Mumbai, Cat.# AL111-500ML) supplemented with 10% fetal bovine serum (CellClone, Genetix Biotech Asia, New Delhi, Cat.# CCS-500-SA-U), 1x penicillin (100U/ml) and streptomycin (100 µg/ml) (Hi-Media, Mumbai Cat. # A018-5X100ML). When 80% confluent, the cell lines were transfected with Cas9-sgRNA vectors (without the donor vector). At 48 h post-transfection, about 10000 of these cells were used for comet assay17 and genomic DNA (gDNA) isolated from the remaining cells was used for T7-endonuclease assay18 to evaluate the in vitro ‘DNA cutting’ activity of Cas9-sgRNA construct. Subsequently, we transfected these cells with the donor vector, Cas9-sgRNA vector and a control vector (pEGFPN1) in the ratio: 2:1:1 using PEI19 from Sigma Aldrich, Inc. (CAS #9002-98-6). After 48 h the cells were imaged and used for downstream applications. Half of the transfected dishes were serially passaged without puromycin selection for two weeks and DNA and protein were isolated. On the remaining dishes puromycin selection was started following the manufacturer’s protocol20 after 36 h of transfection and puromycin resistant colonies at 8 µg/mL were further cultured in puromycin containing media and gDNA was isolated.
Whole-cell extracts (see Cell culture), scraped out and extracted using RIPA Lysis and Extraction Buffer, were run on 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane using a transfer apparatus following the standard protocols (Bio-Rad). After incubation with 5% nonfat milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Tween 20) for 60 min, the membrane was washed once with TBST and incubated with rabbit antibodies against human ABCB11 (Affinity, Catalog #DF 9278) 1: 2000 dilution; human β-actin (Santa Cruz Cat.# SC4778), dilution 1:1000; 4°C overnight. The membrane was washed with TBST buffer and incubated with a 1:5000 dilution of horseradish peroxidase-conjugated anti-rabbit (Santa Cruz Cat# SC-2004)/anti-mouse antibodies (Cat.#SC-2005) for 2 h at room temperature. Blots were washed with TBST four times and developed with the ECL system (Bio-Rad, US Cat.#170-5060) according to the manufacturer’s protocols. Raw, uncropped images from western blotting are available as Underlying data21.
T7 endo I assay detects heteroduplex DNA that results from annealing DNA stands that have been modified after a sgRNA/Cas9 mediated cut to DNA strands without modifications. T7 Endonuclease-1 was purchased from NEB, US (Cat. #NEB #E3321) and was used to digest the PCR products amplified from gDNA extracted from Cas9-sgRNA transfected (test) and un-transfected cells (control) using primers flanking the expected Cas9-sgRNA cut sites following the manufactures protocol18. PCR gel images are available as Underlying data21.
A total of 50–100 cells treated as described in Cell culture were embedded in 0.7% low-melting agarose and mounted on a precoated slide and was immersed in alkaline 0.1% SDS solution overnight, neutralized and electrophoresis was done in an alkaline buffer (pH 10) at 0.74 V/ cm for 30 minutes17. Comet assay images are available as Underlying data21.
Few ampicillin resistant DH5α E. coli colonies which we got after transformation were screened for the insert by colony PCR. One colony was positive for all the fragments of the ABCB11 gene. Sequencing revealed that mutations in ABCB11 (Figure 2). Repeated attempts failed and we considered the possibility of unstable DNA sequences. Therefore, we tried JM109 which gave one positive colony and plasmid was isolated. However, after we soon found the bacteria failing to grow or losing the plasmid on subsequent cultures. Therefore, we transformed One Shot™ Stbl3™ Chemically Competent E. coli cells, which are suitable for cloning unstable DNA segments. We got many positive colonies, however, upon overnight culture the bacteria formed a big pellet (partly lysed bacteria) which cannot be resuspended in phosphate buffered saline. Therefore, we concluded that the ABCB11 gene/gene product is toxic to bacterial cells. It is possible the ABCB11, being a membrane transporter, may be toxic to bacteria. Sequencing files are available as Underlying data21.
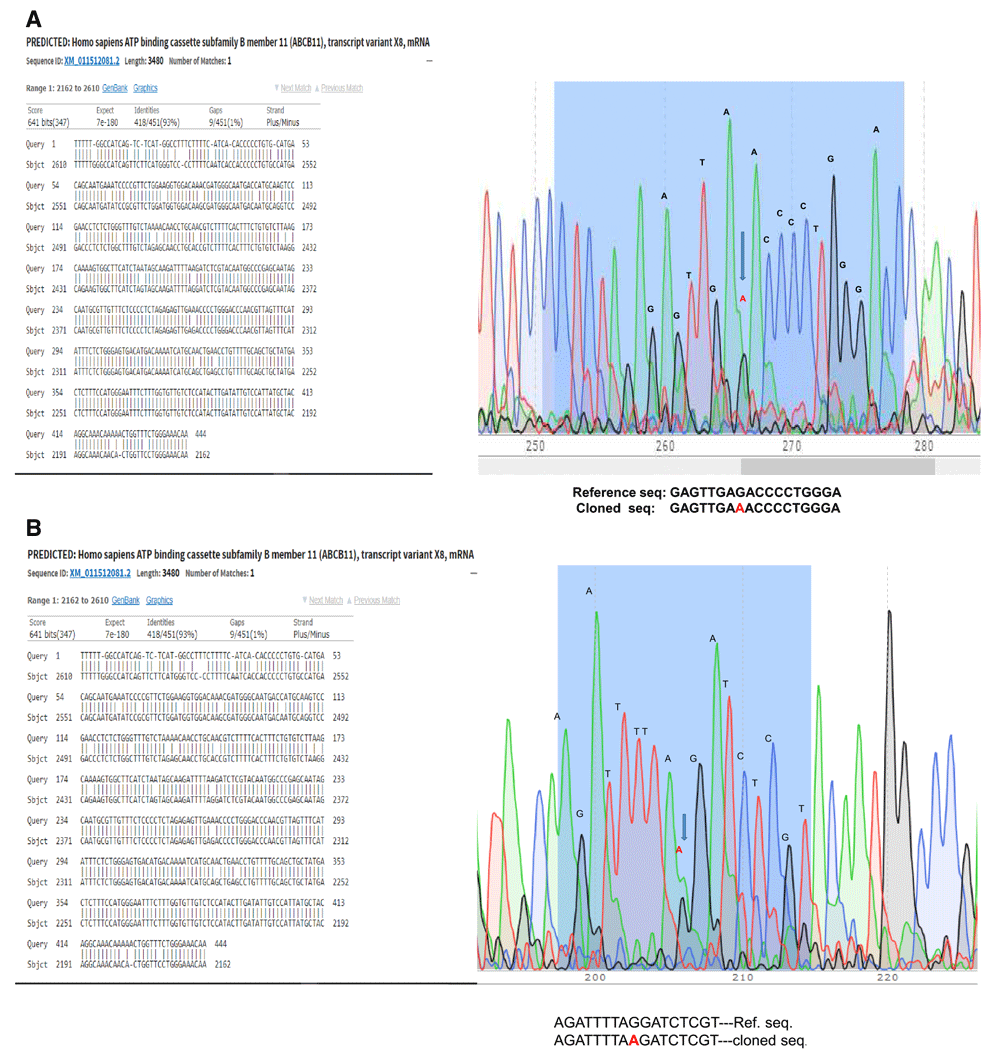
(a) G to A mutation is marked for example. (b) In another example, sequence of another clone, mutation was in a different site.
We conducted PAGE followed by Coomassie staining to see differential protein expression between ABCB11 donor vector transformed bacteria versus untransformed bacteria (Figure 3a; Extended Data)21. We found differential expression of a few proteins. We subsequently performed a western blot and interestingly antibody against human ABCB11 identified a specific protein over expressed in the transformed bacterial cells (Figure 4b). However, in our construct ABCB11 gene was under a eukaryotic promoter. Considering the possibility of some DNA elements which have similarity to bacterial promoters inside the ABCB11 sequence we performed a bioinformatic analysis using BPROM to predict hidden bacterial promoters (Extended data, Supp. Table 2)10. The promoter-site (Pribnow-Schaller box tcatataat) containing sequence (ggttttgagtcagataaatcatataataat) which we identified was modified to (ggtTTCGAAtcagataaatcaTACAACaat) by PCR using an oligonucleotide primer sequence incorporating the modified sequence and subsequent overlap extension PCR amplification of the entire gene fragment. With this modification, we were able to clone ABCB11 coding sequence which was not toxic to bacteria.
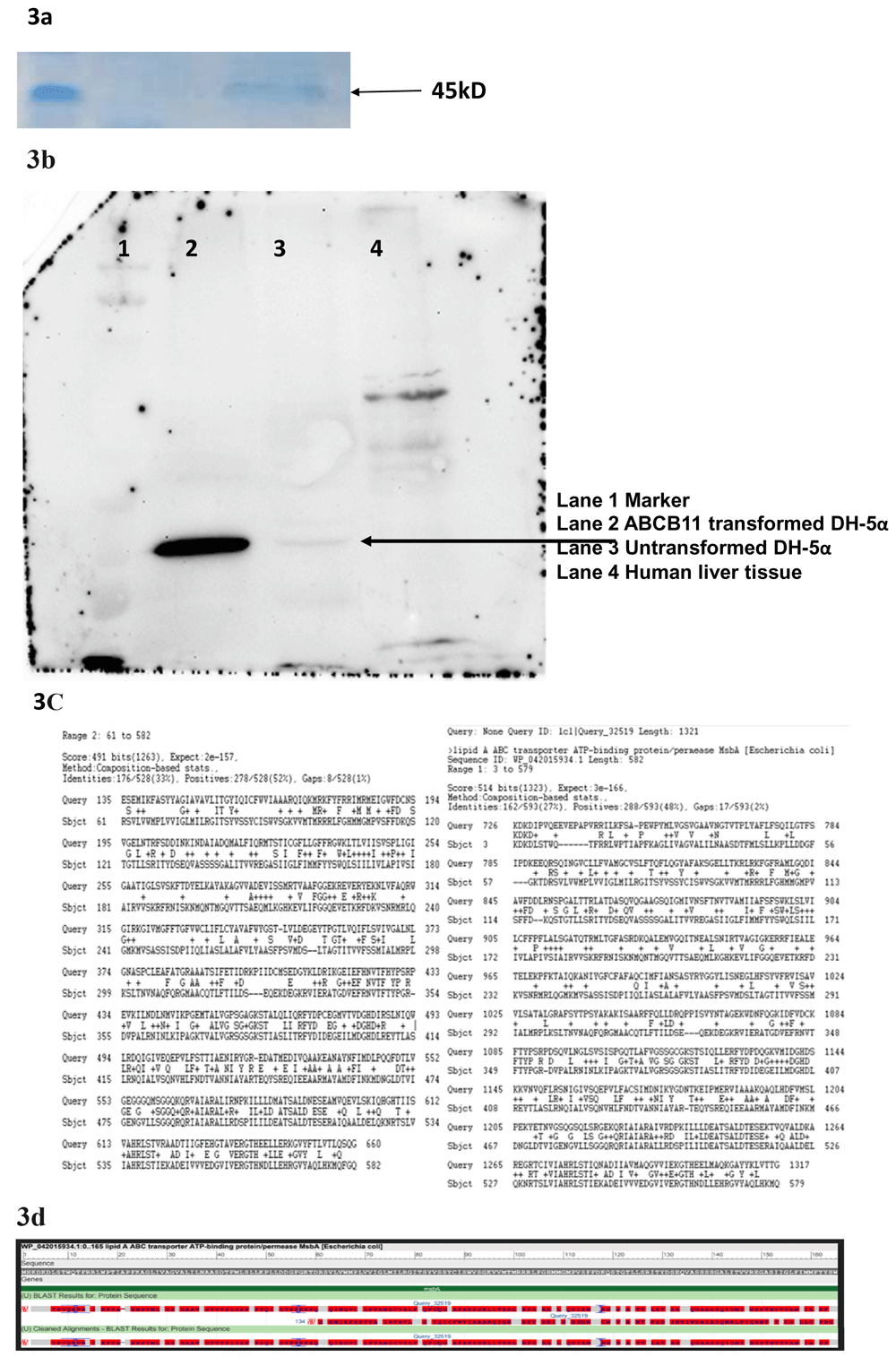
(a) Total protein extract from E. coli transformed the donor vector on Poly Acryl amide Gel Electrophoresis followed by Coomassie blue staining showed differential expression multiple proteins. (b) Western Blot with anti-human ABCB11 antibody shows multiple bands including one probably corresponding to MsbA- a bacterial Lipid A transporter. (c) A bioinformatic analysis (Protein BLAST) revealed ABCB11 and MsbA are sharing conserved domains. (d) Sequence alignment of ABCB11 and MsbA showing conserved domains.
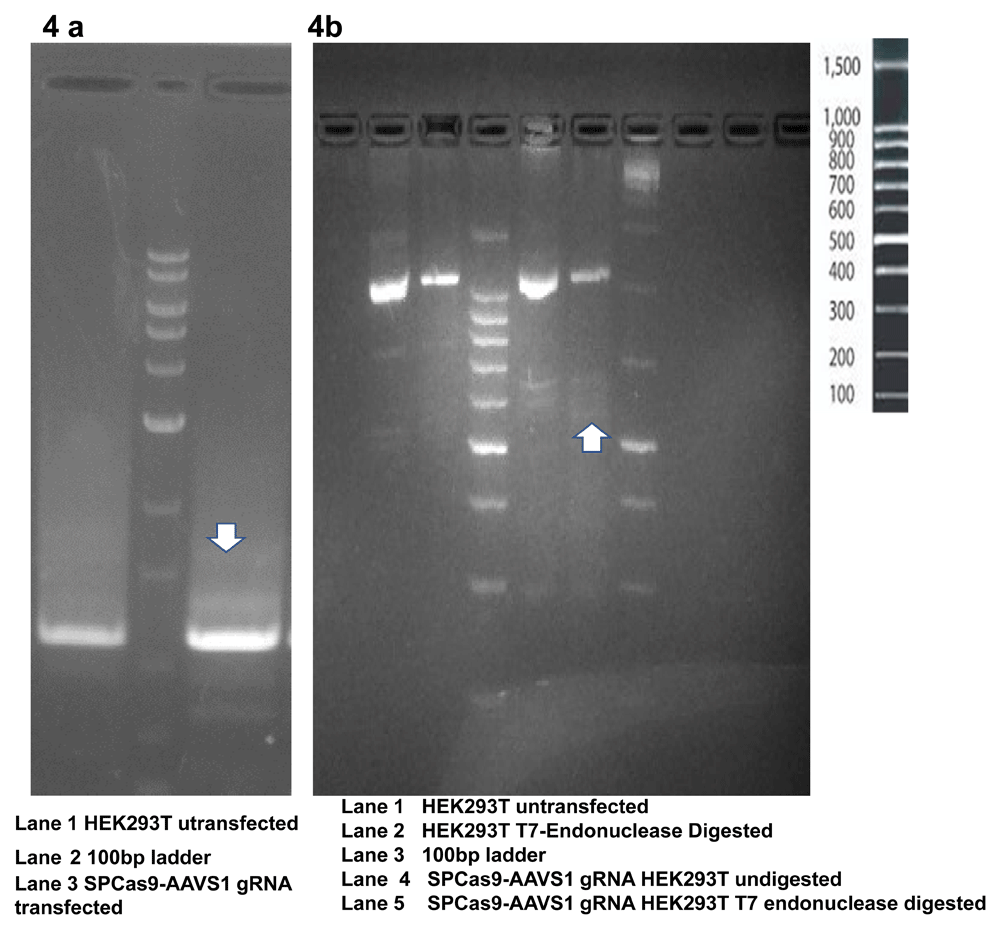
4a The PCR product (even without T7 digestion) shows a distinct band pattern resulting from the formation of heteroduplex. 4b. PCR amplified product after digestion with T7 endonuclease shows faint bands resulting from the digestion of heteroduplexes.
A protein BLAST search identified MsbA (UniProtKB - P60752), a member of the ABC transporter superfamily. MsbA, a 64.46 kD protein, has an important role in E. coli Lipid A (endotoxin or LPS) transport (Figure 3c, d). This protein flips core endotoxin from its site of synthesis on the inner leaflet of the inner membrane to the outer leaflet of the inner membrane. western blot showed identified a unique band in the donor vector transformed E. coli while the untransformed E. coli also showed a faint band but specific band at the same position (Figure 3b).
T7 Endonuclease Assay and Comet Assay were used to evaluate the gDNA cutting activity of Cas9-sgRNA. We observed digestion of heteroduplexes at the CRISPR-Cas9 cut sites which were sensitive to T7 endonuclease (Figure 4a). These heteroduplexes were observed on the agarose gel electrophoresis of PCR products as well (Figure 4b). The Cas9-sgRNA damaged the genome of the transfected cells leading to the formation of comet shaped nuclear material upon electrophoresis (Figure 5).
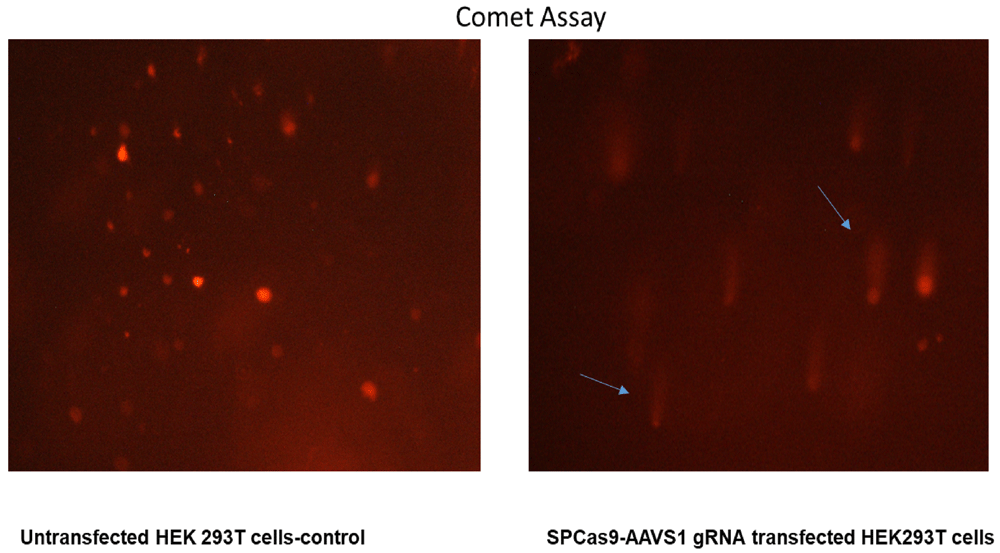
The untreated cells have intact round/oval nuclei while the Cas9-sgRNA treated cells shows a comet shaped nucleus because of DNA damage.
Oligonucleotide PCR primers were designed for bioinformatically predicted off-targets (Extended data, Supp. Tables 3a, b)10. We amplified these segments using genomic DNA extracted from the treated cells (48 h post-transfection with SPCas9-sgRNA vector) as template. The PCR products were sequenced and analyzed for sequence disruption (Table 1).
Oligonucleotide primers designed to amplify these off-target sites to verify off target disruptions.
Western blotting done with total protein extract of fibroblasts 48 h post-transfection with the ABCB11 donor vector showed the expression of ABCB11 protein (Figure 6a). A fibroblast line was used in these experiments because they don’t express ABCB11, naturally while cell lines such as HEK293T and liver cell lines such as HepG2 do express ABCB11.
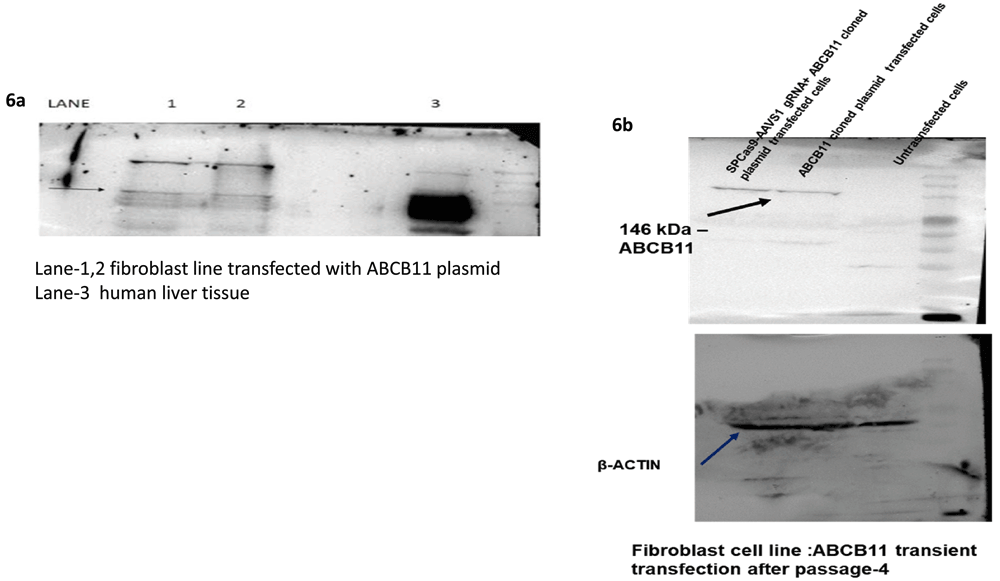
(a) Western Blot with anti-human ABCB11 antibody confirmed the expression after transient transfection with the donor vector having ABCB11. (b) Western Blot was done using total protein isolate from fibroblasts (which does not naturally express ABCB11) after four passages post-co-transfection of the donor vector with ABCB11 gene and the CRISPR-Cas9-sgRNA vector.
Western blotting was repeated with total protein extract from fibroblasts after 2 weeks (fourth passage) post-transfection with SPCas9-sgRNA vector together with the donor vector containing ABCB11. This blot also showed the expression of ABCB11 protein (Figure 6b) suggesting the integration of ABCB11 into the host genome.
We obtained only three puromycin resistant colonies upon transfecting about 20 million HEK cells with a transfection efficiency of 70 to 80% in four 6 cm dishes. The gDNA isolated from transfected cells (Cas9-sgRNA plasmid alone) after 72h, (Cas9-sgRNA plasmid plus donor vector) after 21 days of puromycin selection were used as PCR templates with a forward primer complementary to a region upstream of the 5’ recombination overhang of the vector and a reverse primer complementary to a sequence in the puromycin resistance gene to amplify a segment spanning from a site in the host cell genomic DNA slightly upstream of the genomic integration site to a segment donated by the donor vector (puromycin resistance gene). This PCR product was used as a template for a nested PCR and product was confirmed by restriction enzyme digestion (BamH1) and sequencing (Primers: Extended data, Supp. Table 410, Figure 7). We also PCR-amplified parts of ABCB11 using primers (Extended data, Supp. Table 1)10 which would give specific PCR products from the inserted cassette, to make sure the gene is not deleted from the cassette integrated to the host cell. PCR products showed the expected sizes confirming that the amplified products are from the cassette and not from the native ABCB11 gene present in the cell line (Figure 8).
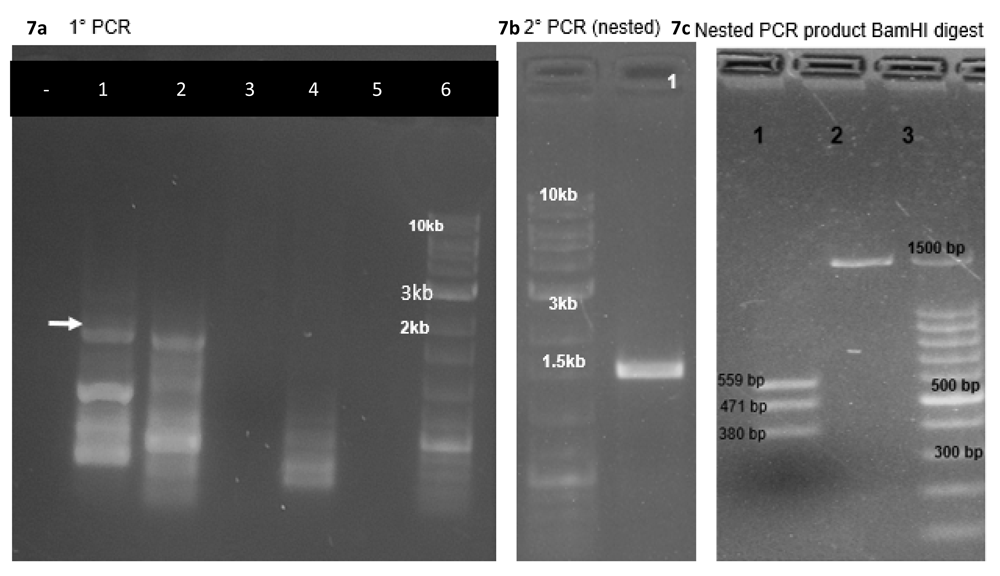
(a) The gDNA isolated from transfected cells (Cas9-sgRNA plasmid alone) after 72h, (Cas9-sgRNA plasmid plus donor vector) after 21 days of puromycin selection were used as PCR templates (lane 1, 2) with a forward primer complementary to a region upstream of the 5’ recombination overhang of the vector and a reverse primer complementary to a sequence in the puromycin resistance gene to amplify a segment spanning from a site in the host cell genomic DNA slightly upstream of the genomic integration site to a segment donated by the donor vector (puromycin resistance gene). Note that genomic DNA from untreated HEK293T cells did not give any products in the expected range (lane 3, 4). (b) The PCR product mentioned in (a) was used as a template for a nested PCR. (c) The PCR product mentioned in (b) was confirmed by restriction enzyme digestion (BamH1) and sequencing (Primers: Extended data, Supp.Table 4)10.

ABCB11-specific primers were used, which would give specific PCR products from the inserted cassette. This was done to make sure the ABCB11 gene was not deleted from the cassette integrated to the host cell genome. The PCR products showed the expected sizes confirming that the amplified products originated from the integrated cassette and not from the native ABCB11 gene present in the cell line.
To our knowledge, this is the first time the ABCB11 gene was inserted into the AAVS1 safe-harbor using CRISPR-Cas9 technology in human cells. Liver directed gene therapy is another approach and was successful in rodents22. Adeno associated vectors do not integrate and therefore the effect of gene therapy many not last in human beings, especially in infants, as the viral vector dilutes out as the cells proliferate in a growing liver23. Another approach is transplantation of hepatocytes differentiated from gene corrected patient iPSC4,24,25. AAVS1 site is considered as a ‘safe haven’ in human genome26 where we chose to insert the gene mutated in PFIC. We could not find any other study which attempted to insert the ABCB11 gene at the AAVS1 site. AAV is a common virus and it is considered non-pathogenic because the seroprevalence of wild-type AAV in humans ranges from 40% for AAV8 to 70% for AAV1 and AAV2, yet; we are not aware of any disease caused by AAV6,7,27. AAV integration into AAVS1 site causes disruption of PPP1R12C (protein phosphatase 1 regulatory subunit 12C). However, this gene is not associated with any disease27. A puromycin gene was placed in the donor cassette such that the puromycin gene will be transcribed only if homologous recombination happens. We obtained only a few puromycin resistant colonies suggesting that homologous recombination was a rare event (~10-7). This suggests that in vivo gene therapy using CRISPR-Cas9 technology making use of homology directed gene repair could be difficult. “Targeted Integration and high transgene expression in AAVS1 Transgenic mice after in-vivo hematopoietic stem cell transduction with HDAd5/35++ Vectors”28 is reported; however, to achieve this they used an adenoviral gene delivery system with AAV5 ITRs and AAV35 helper. Integration of AAV/ Cas9 into Cas9 mediated cut sites is a potentially hazardous consequence of this approach29,30.
We found that the human ABCB11 donor vector transformed bacteria either died or the ABCB11 gene sequence got mutated meaning either the DNA sequence or the ABCB11 protein has some untoward effects on bacteria. We performed a western blot and found that in ABCB11-transformed bacterial clones giving a band around 45 kD and HepG2 cells/liver tissue is giving a band at around 140 kD which corresponds to ABCB11. It was interesting to note that untransformed E. coli cells are also showing a band although very faint around 45 kD which suggested the possibility of a bacterial protein which might have structural similarity to human ABCB11. We performed a bioinformatic search and identified MsbA an E. coli protein which functions as a lipid transporter (~64 kD). MsbA is involved in the transport of bacterial endotoxin-a function like the ABCB11 which transports the bile salts which are lipid derivatives31,32. Another interesting observation was the identification of a bacterial promoter sequence (Pribnow-Schaller Box) in human ABCB11 causing unexpected expression of ABCB11 protein and bacterial toxicity (Figure 9). This was an important lesson for us because we spent a lot of time trying to clone ABCB11. It is therefore important to search for and eliminate if any bacterial promoter sequences or similar elements are identified, for successful cloning of eukaryotic/toxic genes in E. coli. We do not know how exactly ABCB11 caused bacterial toxicity. Possibly, expression of ABCB11 in E. coli might be destabilizing the E. coli membrane, since ABCB11, being a lipid transporter, is a membrane-spanning protein.
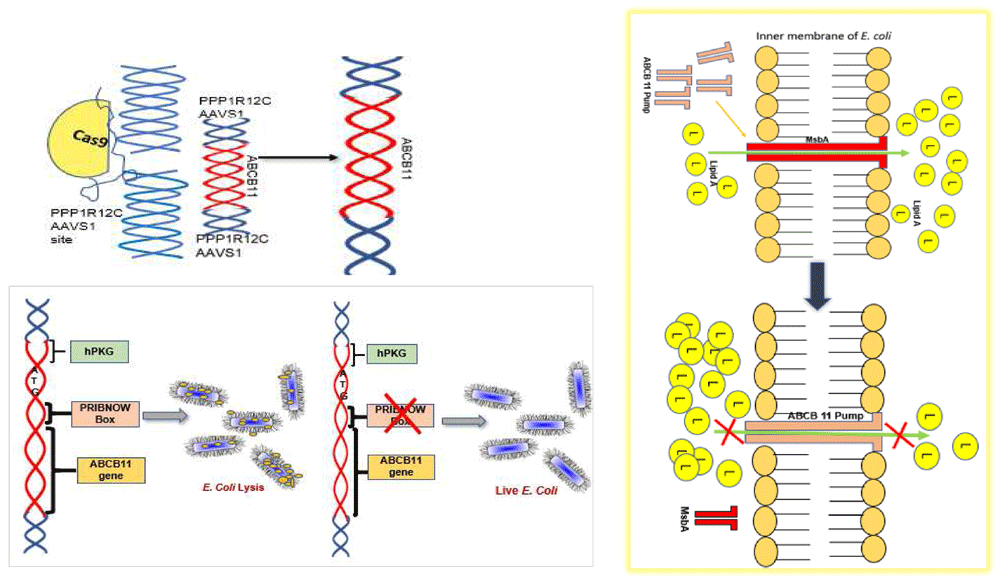
A Pribnow box was detected inside ABCB11, which allowed the gene to transcribe in E. coli, causing bacterial lysis, probably through competitive replacement of a homologous transporter protein in E. coli (E. coli Endotoxin (Lipid A) Transporter) MsbA, resulting in Lipid A (L) accumulation inside the bacteria.
Alternatively, the human protein, which is similar to the E. coli protein might have caused a competition between the bacterial transporter MsbA and human protein for membrane incorporation resulting in the accumulation of endotoxin within E. coli cells because unlike the bacterial transporter the membrane incorporated ABCB11 might not be able to transport endotoxin out. This raises the possibility that endotoxin is toxic to E. coli itself if it is not exported and MsbA can therefore be considered as a drug target. This point required further validation (Figure 9). The advantage of such an antibiotic is that it will be selective to endotoxin producing microbes. More research is required in this direction. It may be noted that endotoxin producing microbes play an important role in sepsis33 and diseases such as non-cirrhotic portal hypertension34–36.
To conclude, we successfully inserted ABCB11 at the AAVS1 site using CRISPR-Cas9, however, the frequency of homologous recombination was very low as evident from the number of puromycin resistant colonies. With this low efficiency, with the current technology it is unlikely that this approach would be successful in in-vivo gene editing. It is worth, exploring MsBA as a novel antibiotic target for LPS producing bacteria, although our data in this direction is primitive and requires further validation.
Harvard Dataverse: Sequencing Data, supplementary data to Cloning of Human ABCB11 Gene in E. coli required the removal of an Intragenic Pribnow-Schaller Box before it’s Insertion into Genomic Safe Harbor AAVS1 Site using CRISPR Cas9. https://doi.org/10.7910/DVN/32TXCD21.
This project contains the following underlying data:
2020_06_20_211607 actin.jpg. (Uncropped western blot image.)
4a .T7 endonuclease digestion.jpg. (PCR gel image.)
4b. PCR without T7 endonuclease.jpg (PCR gel image.)
4c... Cas9-sgRNA treated cells comet assay.jpg. (Image taken from Comet assay, treated cells.)
4c...... untreated cells comet-1.jpg. (Image taken from Comet assay, untreated cells.)
ABCB11_fibroblasts.jpg. (Uncropped western blot image.)
ABCB11_fragments.tif. (PCR gel image.)
ABCB11_WB_P4_2020_06_19_183004-1.tif. (Uncropped western blot image.)
DH5a WB-ABCB11-long_Exposure.jpg. (Uncropped western blot image.)
DH5a WB-ABCB11.jpg. (Uncropped western blot image.)
E.Coli_PAGE_Coumasse.jpg. (Uncropped PAGE gel.)
JM109_ABCB11 WB.tif. (Uncropped western blot image.)
nested PCR product BamH1 digest.jpg. (PCR gel image.)
Nested PCR secondary.jpg. (PCR gel image.)
Nested_Primary pcr.jpg. (PCR gel image.)
Repeat_WB_2020_07_11_181831.jpg. (Uncropped western blot image.)
Sequencing data.7z. (Sequencing data produced in the present study.)
T7endo 30420202.jpg. (PCR gel image.)
Harvard Dataverse: Supplementary Tables to Cloning of Human ABCB11 Gene in E. coli required the removal of an Intragenic Pribnow-Schaller Box before it’s Insertion into Genomic Safe Harbor AAVS1 Site using CRISPR Cas9. https://doi.org/10.7910/DVN/NTUOXM10.
This project contains the following extended data:
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)