Keywords
CAKUT, kidney development, duplex systems, ureter budding
CAKUT, kidney development, duplex systems, ureter budding
The urinary tract, composed of the kidneys, ureters, bladder and urethra, represents the main excretory system of the mammalian organism. Development of the urinary system, made up of more than 40 different cell types, needs to proceed in a highly organised manner. Given this complexity, it is not surprising that mutations in developmental genes can lead to a wide variety of abnormalities that are usually grouped together as congenital abnormalities of the kidneys and urinary tract (CAKUT). Defects affecting the kidneys range from renal agenesis (a complete lack of kidney development) to hypoplasia (reduced size), dysplasia (abnormally developed tissue), cystic dysplasia, and terminal differentiation defects. Lower urinary tract malformations include vesicoureteral reflux (VUR), hypospadias (opening of the urethra at the lower side of the penis) and posterior urethral valves that often lead to outflow obstructions. Although individual malformations are considered rare diseases, CAKUT, taken together, have an incidence of about 3 to 6 in 1000 live births and thus belong to the most frequent abnormalities detected in the new-born child1. An in-depth presentation of all subclasses and their aetiology would be far beyond the scope of this review and therefore interested readers are referred to other publications that present an overview of CAKUT phenotypes and the genetics underlying them2–6. Here, we will instead concentrate on duplex (or multiplex) kidneys, a very frequent subclass of CAKUT, which is often neglected in the literature.
To understand the aetiology of duplex kidneys, it is important to consider how the urinary system forms. From a developmental point of view, the urogenital tract derives from two independent germ layers with kidneys and ureters arising from the intermediate mesoderm (IM) and the bladder and urethra developing from cloacal endoderm7. Accordingly, malformations of the urinary system can be further classified into congenital abnormalities of the upper and lower urinary tract (and the latter are sometimes abbreviated as CALUT). Despite this developmental distinction, it should be noted that some authors group malformations of the ureter as part of congenital abnormalities of the lower urinary tract.
Kidney development in mammals commences with the formation of the nephric duct (ND) at the anterior (rostral) pole of the IM. As development proceeds, epithelial cells of the ND proliferate and actively migrate towards the caudal end of the nephrogenic cord8–10. Eventually, the ND fuses with the cloaca, a process that involves dedicated apoptosis and requires GATA3 and LHX1 as well as retinoic acid and RET and FGF signalling9–13.
As the ND elongates caudally, a series of tubules forms within the nephrogenic cord. The most anteriorly positioned pronephric tubules are considered an evolutionary remnant and are non-functional in mammals. Subsequently, a wave of mesonephric tubules develop that fall into two groups. While rostrally positioned tubules are connected to the ND and serve as an embryonic kidney, more caudally located tubules do not drain into the ND and are non-functional14,15. Both pronephros and mesonephros are transitory structures in the mammalian embryo and disappear (pronephros) or are remodelled (mesonephros) at later stages of development.
The metanephros represents the permanent kidney in mammals and develops at the most caudal position of the IM. Metanephros development is first detectable as a population of slightly condensed mesenchymal cells within the nephrogenic cord which express a set of molecular markers (HOX11, SIX2, GDNF, EYA1)15,16. In normal development, signalling from the metanephric mesenchyme (MM) induces the formation and outgrowth of a single ureteric bud (UB) from the ND, which will invade the MM and undergo a first stereotypic dichotomous branching event (T-shaped ureter). The collecting duct system (ureteric tree) forms through further rounds of branching that often include tri-tips, which, however, eventually resolve into ureter bifurcations17,18. In return, signals released from the ureter will induce the MM to differentiate into nephrons, the functional units of the kidney. For further details on this process, we refer the reader to recent reviews19–22.
Development of the urinary system is not restricted to kidney formation but also involves extensive developmental remodelling of the lower tract. An excellent and detailed description of this complex process can be found in 7. In brief, the emerging UB is initially connected to the cloaca via the distal part of the ND, also termed the common nephric duct (CND). Downgrowth of the urorectal septum leads to a separation of the cloaca into a ventrally located urogenital sinus and a dorsally positioned anorectal sinus7,23–25. The cranial urogenital sinus will further elongate to develop into the bladder, whereas its posterior portion will form the urethra. As development proceeds, apoptosis eliminates the CND, leading to the fusion of the ureter with the future bladder, thus creating the ureterovesical junction26.
Duplex systems can have a variety of phenotypes, and multiple classification systems have been proposed to categorise this pathology (Figure 1)27. In incomplete duplication, the two poles of a duplex kidney share the same ureteral orifice of the bladder. Such duplex kidneys with a bifid pelvis or ureter arise when an initially single UB bifurcates before it reaches the ampulla. This is likely caused by a premature first branching event that occurred before the ureter has reached the MM. Much more frequent are complete duplications, which occur when two UBs emerge from the ND. In most cases, the lower pole of the kidney is normal and the upper pole is abnormal28,29, an observation explained by the fact that the ectopic UB frequently emerges anteriorly to the position of the normal UB and drives the formation of the upper pole of a duplex kidney. Inverted Y-ureteral duplication is a rare condition in which two ureteral orifices drain from a single normal kidney. Inverted Y-ureteral duplication is believed to be caused by the merging of two independent UBs just before or as they reach the kidney anlagen30. A very rare H-shaped ureter has also been reported31. Although the vast majority of cases involve a simple duplication, multiplex ureters with up to six independent buds have also been described32–37. In some cases, the additional ureter or ureters are ectopic and fail to connect to the bladder or the kidney (blind ending ureter)33.
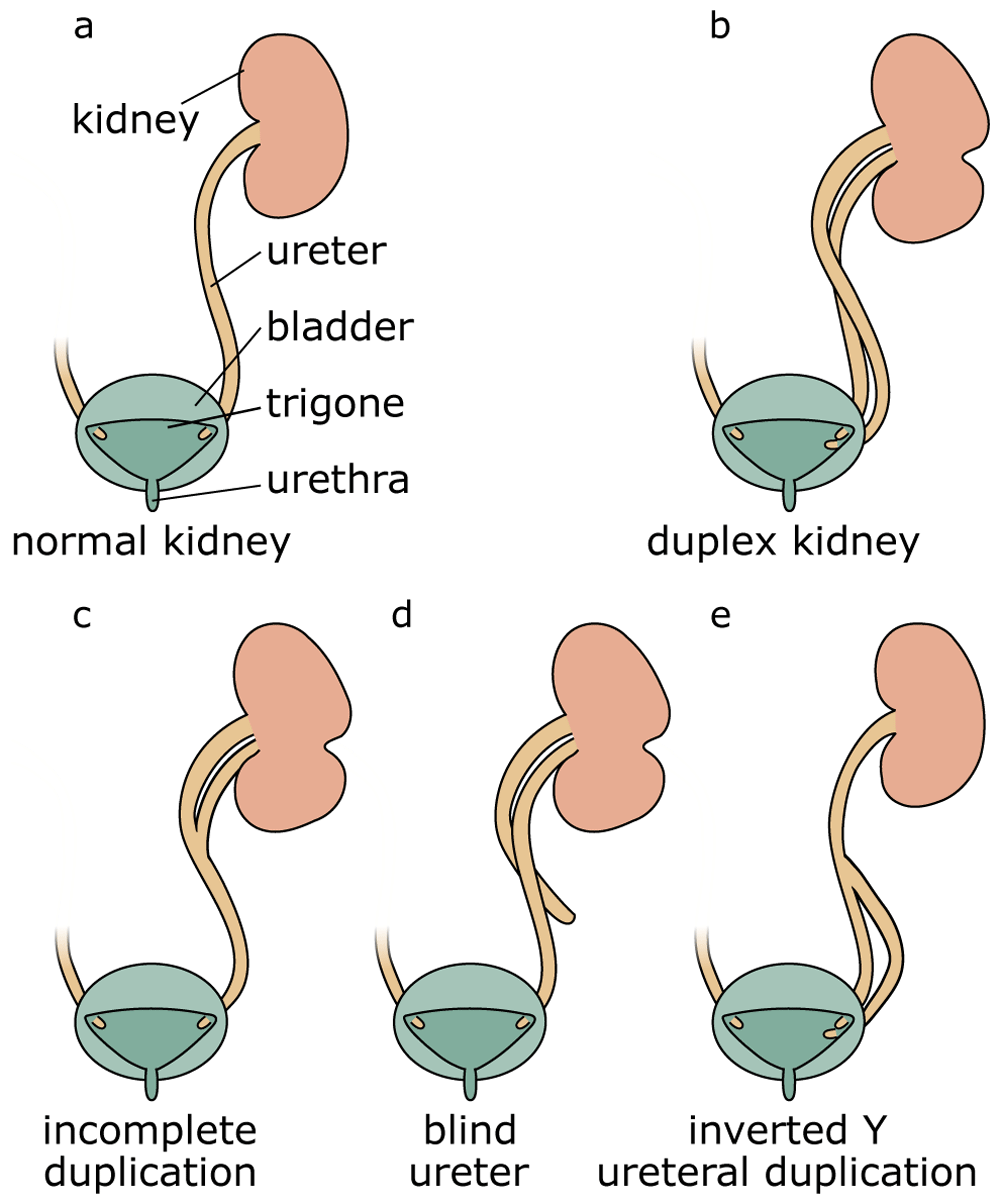
Compared with a normal kidney (a), complete duplication produces a duplex kidney with two poles that drain into two ureters (b). Incomplete duplication leads to a Y-shaped ureter (c). Blind ureters do not drain into the bladder (d). In the rare case of inverted Y-ureteral duplication, two ureters fuse before entering the kidney (e).
The aetiology of most duplex kidneys can be traced back to the very first induction steps of the ureter. In the majority of cases, an additional UB emerges in a rostral position to the normal outgrowth. By contrast, in adults, the upper (abnormal) kidney pole drains into the bladder at a site distal to the orifice of the lower kidney pole38. This paradoxical phenomenon, known as the Weigert–Meyer rule29, can be explained by the significant amount of remodelling occurring at the future ureter–bladder junction during development. Indeed, as apoptosis eliminates the CND, the ureter inserts into the developing bladder and moves upwards (Figure 2)7,39–41. An initially anteriorly positioned ureter thus ends up with a more distal insertion site in the bladder, a model that has been proposed by Mackie and Stephens42. Correct positioning of the ureter into the bladder is important to allow formation of a normal trigone (the triangle formed by the two ureter orifices and the urethra) and prevent ureter reflux caused by a malfunctioning valve or a too-short ureter tunnel. Because the vast majority of duplex kidneys arise from an ectopic bud in a rostral position, it is usually the upper pole of the kidney that is affected by VUR and hydronephrosis.
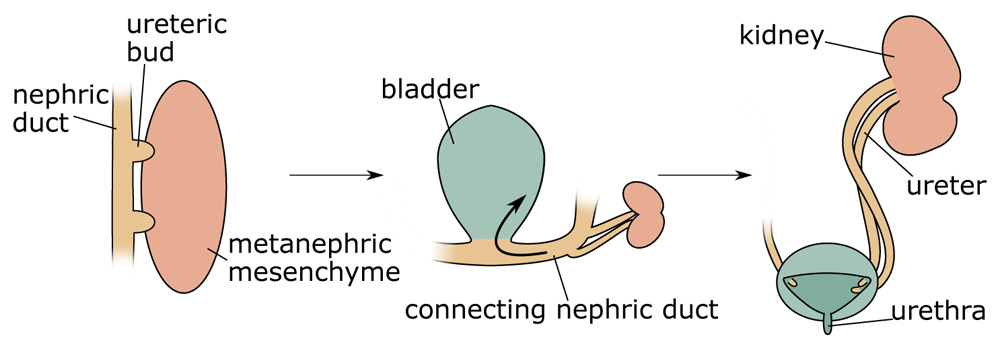
Duplex kidneys form through the induction of two ureteric buds from the nephric duct that will invade the metanephric mesenchyme. Subsequently, apoptosis of the common nephric duct (CND) leads to the insertion of both ureters into the developing bladder with the orifice of the initially posteriorly positioned ureteric bud ending up in a superior position.
Estimates suggest a prevalence of duplex kidneys of between 0.2 and 2% in the general population, and females are affected twice as frequently as males38,43. The reasons for this sex bias are unknown. About 40% of patients with duplex kidneys have been reported to exhibit pathological manifestations43. However, because duplex kidneys are frequently asymptomatic and therefore predominantly detected in patients who seek medical assistance, the actual percentage of patients with symptoms is likely to be lower. Symptoms associated with duplex kidneys can include pain, haematuria, dysuria and difficulty or abnormal frequency of micturition38,43. Specific manifestation of the pathology depends on the anatomy of each duplication event44. Furthermore, duplex kidneys are linked to a number of renal disorders, including pelvi-calyceal dilatation, cortical scarring, VUR, hydronephrosis, ureterocoeles on the non-duplex side, caliculi or yo-yo reflux (in the incomplete duplication cases)38,43.
If duplex kidney formation is rooted in the formation of two ureteric tips, how can we explain the outgrowth of supernumerary buds on a molecular level? Interactions between MM and the ND are crucial to ensure the induction of the ureter from the ND, and a key pathway controlling this process is the GDNF/RET signalling axis (Figure 3)45. GDNF, a distant member of the transforming growth factor beta (TGFβ) superfamily of signalling molecules, is specifically expressed within the MM, whereas its cognate receptor RET is expressed along the entire length of the ND. Binding of GDNF to RET is greatly facilitated by the co-receptor GFRα1. The requirement for these genes in ureter outgrowth has been extensively demonstrated by using gene targeting in mice, and homozygous mutations in either of these genes leads to a failure of ureter induction and consequently renal agenesis46–50. Binding of GDNF to the receptor tyrosine kinase RET induces autophosphorylation and recruitment of the tyrosine phosphatase SHP251,52, which results in the activation of several intracellular signalling cascades, including RAS/MAPK, PLCγ/Ca2+, PI3K-AKT53, and culminates in the transcriptional activation of a set of downstream target genes54. Ureter branching appears to involve, in particular, the ERK/MAPK pathway, and mice lacking the kinases Mek1 and Mek2 fail to form a properly branched ureteric tree55. Activated RET signalling induces not only proliferation but also cellular motility. Indeed, experiments in chimeric mice demonstrated that wild-type cells move towards the tip of the UB but that Ret mutant cells are left behind56. This cellular sorting mechanism ensures a strong and directed response that, under normal circumstances, results in the outgrowth of a single UB.
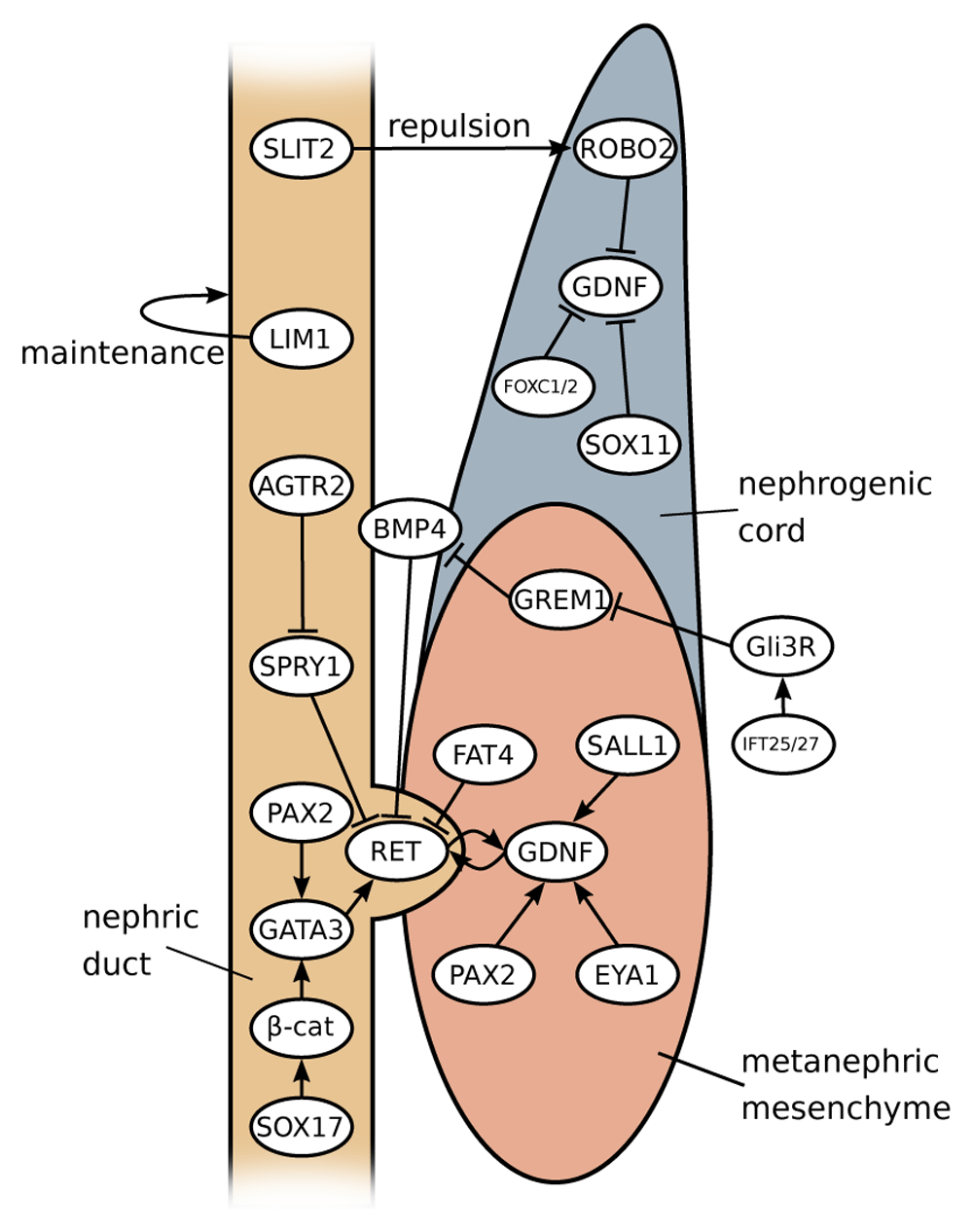
GDNF-RET signalling is at the core of the signalling network in kidney development and is responsible for ureteric bud (UB) emergence. GDNF expression is positively modulated by factors expressed in metanephric mesenchyme (PAX2, EYA1 and SALL1) and negatively (probably in an indirect manner) by SOX11, FOXC1, FOXC2 and ROBO2 in anterior domains of the nephrogenic cord. Ectopic formation of the UB is prevented by the factors expressed in nephric duct (SLIT2, SPRY1 and GATA3) and in the enveloping mesenchyme (BMP4 and FAT4). Influence from other upstream factors leads to the formation of a complex regulatory landscape.
Given the crucial function of Gdnf in ureter induction, we need to consider how the expression of this gene is regulated. Activation of Gdnf in the mesenchyme relies on a set of transcription factors, including SALL1, PAX2 and EYA1. Deletion of either of these factors in mice leads to a lack of ureter induction and consequently to renal agenesis57–59. Heterozygous mutations in each of these genes have been shown to be involved in CAKUT5,6, and SALL1, in particular, has also been linked to duplex kidney formation60. In addition, Gdnf mRNA levels appear to be regulated post-transcriptionally via its 3’ untranslated region (UTR). Indeed, replacement with a heterologous UTR sequence resulted in increased Gdnf expression levels that were associated with ND remodelling defects independent of apoptosis61.
In the mouse, Gdnf expression commences in rostral domains of the nephrogenic cord at embryonic day 9.5 (E9.5), about 1 day before ureter induction. The ND, however, does not respond to the GDNF signal in those anterior regions and this is likely to be due to two reasons: First, the anterior IM has relatively high levels of BMP signalling, which is known to suppress ureter branching (see the ‘Restricting Ret activation’ section below). Second, SLIT/ROBO signalling, a pathway that is known for its role in axon repulsion62, appears to repulse Gdnf-expressing cells from the ND, thus causing a physical separation of these two structures in anterior regions63. Robo2 knockout mice lack this separation and show ectopic buds along the entire length of the ND64. The physical separation of ND and Gdnf-expressing cells may explain why in Foxc1, Foxc2 and Sox11 mouse mutants, which all display a dramatic expansion of Gdnf expression, the ND does not respond in the anterior domain64–66. Instead, only the region just rostrally to the normal site of induction responds by forming a second ureter. Mutations in ROBO2, SLIT2 and its associated GTPase-activating protein SRGAP2 are found in patients with VUR and duplex systems67,68.
By the time of ureter induction (E10.5 in mice), mesenchymal cells that express Gdnf become restricted to the caudal part of the MM (Figure 4). Three possible mechanisms for this restriction could be envisaged: (1) Active suppression of Gdnf expression in more rostral domains could occur. Since the expression of Foxc1 and Sox11 overlaps with the Gdnf domain, active suppression of the latter seems unlikely. (2) Gdnf-positive cells at the rostral end could undergo cell death. The pro- and mesonephros are known to be subject to massive apoptosis, although this seems to affect, in particular, the epithelial cells of tubules. However, preliminary data from our lab suggest that mesenchymal cells positioned just rostrally of the Gdnf-expressing domain also undergo apoptosis. (3) Finally, rostrally positioned Gdnf-positive cells may undergo directed migration towards the caudal end. The proposed distinct origin of ND and MM from the anterior and posterior IM, respectively, would argue against this possibility16. However, Slit/Robo signalling and members of the SoxC class genes have been implicated in cell migration62,69. Perhaps Gdnf restriction is a combination of several mechanisms, including cell clearance through apoptosis and directed cell migration of anteriorly positioned Gdnf-positive cells. A careful analysis of mouse mutants showing an expansion of the Gdnf expression domain, perhaps coupled with live imaging in explant cultures, may help to address this open question.
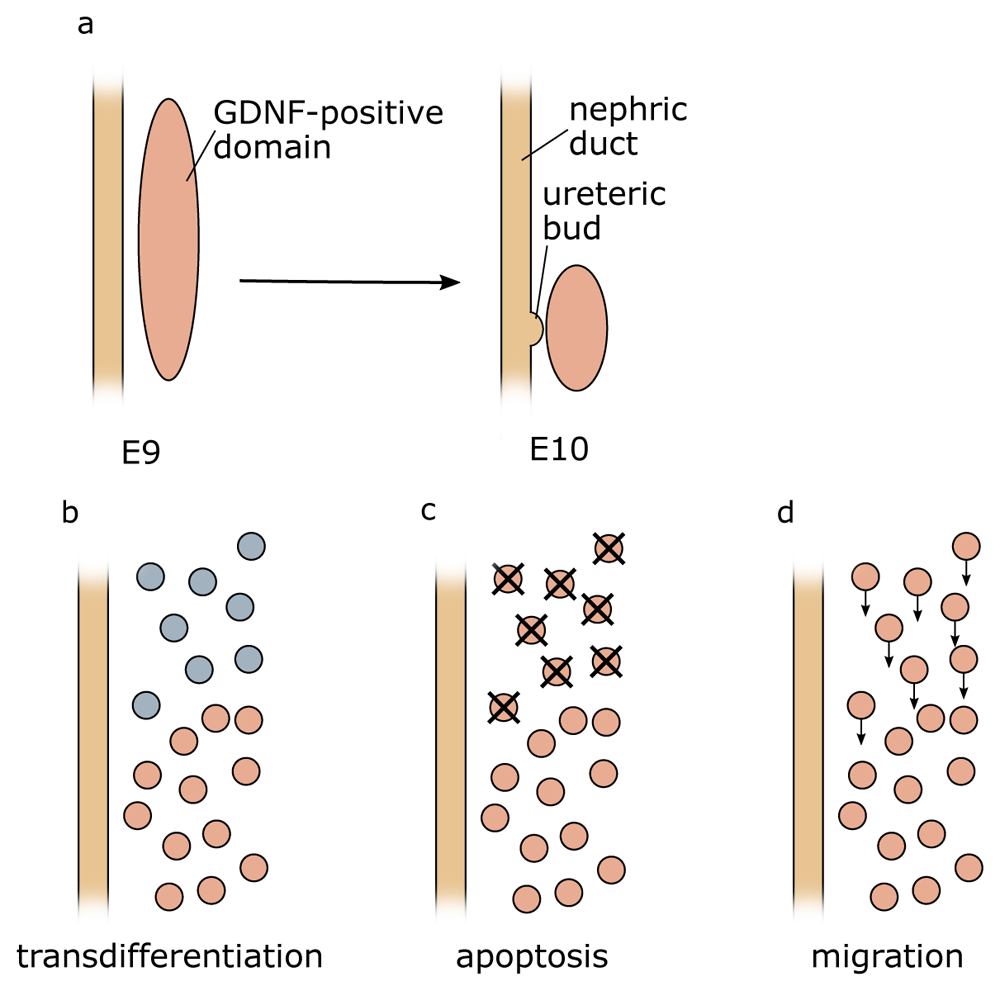
(a) At early stages during development, Gdnf expression can be found in rostral domains of the intermediate mesoderm but over time becomes caudally restricted. Three mechanisms could explain this observation: (b) active suppression of Gdnf expression in more rostral domains (c), apoptosis of Gdnf-expressing cells (d), or migration of the cells towards the caudal end of the intermediate mesoderm. E, embryonic day.
PAX2 not only is involved in the activation of GDNF but also is required for the expression of ND-specific genes. A key target appears to be the transcription factor GATA3, which in turn transcriptionally activates Ret. Tissue-specific knockout mice that lack Gata3 within the ND show an altered response to local growth factors (GDNF and FGF) and display premature cell differentiation and differential cell adhesion properties. As a result, cells with sufficient levels of GATA3 and RET segregate from GATA3-deficient cells and expand, forming ectopic buds and kidneys70.
Beta-catenin, a multifunctional protein involved in cell–cell adhesion and transcriptional regulation, appears to be one of the factors involved in this growth. Conditional inactivation of β-catenin in the ND leads to a range of kidney defects, including duplex kidney formation71. Molecular markers affected in these mutants were the transcription factors EMX2 and SOX9, both of which are known to be involved in ureter budding72,73. However, ectopic budding was observed only in cases where loss of β-catenin expression was mosaic74. Hypoxia-induced reduction of β-catenin has also been shown to cause duplex kidneys amongst other CAKUT phenotypes75. Beta-catenin action during kidney induction is mediated at least partly through the transcription factor GATA370.
Sox17 mutations have been identified in a cohort of human patients with CAKUT, including a duplicated pyeloureteral system. The authors demonstrated that the mutation influenced protein stability and reduced β-catenin activity76. It is therefore possible that the mutated SOX17 protein leads to lower β-catenin and, in turn, reduced GATA3 levels.
In parallel to GATA3, LHX1 (LIM1) appears to be essential in permitting normal budding77. Tissue-specific deletion of LIM1 in ND derivatives leads to renal hypoplasia and hydronephrosis and an impaired extension of the ND. Some conditional mutants of Lim1 also display incomplete duplication of kidney ureters where both poles of a duplex kidney merge before entering the bladder. This form of duplex kidney was traced back to the first UB branching event, where defective UB forms a Y-shaped rather than a T-shaped structure9.
To limit ureter outgrowth to a single site, a series of negative regulators that suppress the RET signalling cascade are in place. BMP signalling, in particular, seems to be a suppressor of ureter outgrowth and branching, and heterozygous Bmp4 mutations in mice lead to a wide range of CAKUT phenotypes, including duplex kidneys78. BMP and FGF signalling are known antagonists in epithelial branching of the lung79 but also kidney development80. Since FGF and RET receptors are receptor tyrosine kinases that use similar intracellular signal transduction pathways, we can reason that the antagonistic action of BMP acts in analogous fashion on RET signalling. To permit ureter outgrowth specifically at the site of the future kidney, MM cells express the BMP inhibitor Gremlin (Grem1), which counteracts the BMP function81. Heterozygous BMP4 and GREM1 mutations have both been identified in human patients with CAKUT82,83, although it is not clear whether variants in these genes also predispose to duplex kidney formation.
A number of other genes involved in duplex kidney formation appear to affect the BMP/Gremlin axis. Mutants for the intraflagellar transport proteins IFT25 or IFT27, which are believed to increase GLI3R, a repressor of SHH signalling, show a high penetrance of duplex kidney formation (~50%)84. Similarly, constitutive expression of a truncation mutation in Gli3 (Gli3Δ699), which is found in Pallister–Hall syndrome and is likely to sensitise tissue for SHH signalling, causes CAKUT with duplex kidneys85. The phenotype has been linked to an increased sensitivity of the ND by lowering BMP4 signalling.
Of interest, several genes that are implicated in the formation of cilia (for example, Cep290, Dync2h1, Tbc1d32 and Tmem67) have also been implicated in duplex kidney formation86. The primary cilia is an organelle that has a key function in cellular signalling87, and SHH signalling, in particular, is directly linked to this organelle. Because SHH signalling has been proposed to be involved in duplex kidney formation (88 and above), it is tempting to speculate that the above cilia-related genes also influence this pathway.
In addition to extracellular modulators, cytoplasmic antagonists exist to suppress ureter outgrowth. Most notably, Sprouty (Spry1) suppresses MAPK signalling in the absence of GDNF, and inactivation in mice results in the formation of multiple UBs89. Signalling through the angiotensin receptor appears to be important in suppressing Spry1 expression90 but also in activating Ret expression, and Agtr2 knockout mice show a range of CAKUT phenotypes, including a duplex system91. To date, no pathogenetic SPRY1 mutations have been identified in patients with CAKUT, and it is currently unclear to what extent this gene contributes to duplex kidney formation in human patients. Interestingly, in the absence of Spry1, GDNF signalling is no longer required for ureter induction, and Spry1-/-/Gdnf-/- double knockout mice develop normal kidneys. In this context, FGF10, which normally plays only a minor role in kidney development, becomes indispensable for kidney induction, and triple mutants (Fgf10-/-/Spry1-/-/Gdnf-/-) display renal agenesis92. FGF signalling thus can be considered a reinforcing signal that contributes to enhanced epithelial growth and budding. FGF signalling serves as the main pathway in branching morphogenesis of other organs such as the lung93, and we can speculate that GDNF/RET signalling has taken over the ancestral function of FGF in epithelial branching of the kidney.
Finally, tissue-specific knockout of Fat4 within the nephrogenic cord results in a duplex kidney phenotype that can be rescued by reducing the dose of GDNF (Gdnf+/-). Recent molecular experiments demonstrated that FAT4 directly binds to RET and restricts its activity in the ND/UB by disrupting the formation of RET-GFRA1-GDNF complex94,95.
There are a number of other genes which have been shown to be implicated in duplex kidney formation but for which the molecular events leading to supernumerary buds are not well defined. Because in these cases the causative nature of mutations for duplex kidney formation is less established, we refrain from a mere listing of genes at this place. The interested reader is referred to Table 1 of genes involved, the associated phenotypes and corresponding references.
| Group | Genotype | Mechanism | Reference |
|---|---|---|---|
| GDNF domain | |||
| Robo2-/- | Abnormal Gdnf expression domain MM fails to separate from WD | Grieshammer et al.64 Wainwright et al.63 | |
| Slit2-/- | Abnormal Gdnf expression domain | Grieshammer et al.64 | |
| Foxc1-/- | MM fails to reduce in size | Kume et al.65 Komaki et al.96 | |
| Sox11-/- | MM fails to reduce in size | Neirijnck et al.66 | |
| Increased sensitivity of WD | |||
| Bmp4+/- | Lack of inhibition of WNT11, a target of GDNF | Miyazaki et al.78 Michos et al.81 | |
| Ift25-/-, Ift27-/- | Increased sensitivity of WD through Gremlin-BMP4 cascade | Desai et al.84 | |
| Gli3Δ699/Δ699 | Increased sensitivity of WD through Gremlin-BMP4 cascade | Blake et al.85 | |
| Agtr2-/Y | Disrupted renin-angiotensin signalling leads to aberrant UB morphogenesis | Nishimura et al.91 Yosypiv et al.90 | |
| p53-/-, p53UB-/- | Increased response of WD to GDNF signal. Two ureters fuse in the later development and resemble a bifurcation | Saifudeen et al.97 El-dahr et al.98 | |
| Fat4-/- Fjx1-/- | Premature branching with incomplete duplication due to overactive GDNF-RET signalling | Saburi et al.94 Zhang et al.95 | |
| Hoxb7-Cre β-catenin-/c | Ectopic activation of UB branching pathway in WD | Marose et al.74 | |
| Spry1-/- | Increased sensitivity of WD to GDNF-RET signalling | Basson et al.89 | |
| Gata3ND-/- | The entire length on WD is covered by ectopic UBs, most of which subsequently regress | Grote et al.70 | |
| Cell polarity defect | |||
| T-Cre Wnt5afl/Δ | Double UB, abnormal morphology of posterior WD, defects in IM morphogenesis | Yun et al.99 | |
| Ror2-/- | Similar to Wnt5a phenotype | Yun et al.99 | |
| Cell adhesion defect | |||
| L1-/Y | Either incomplete or complete duplication. Double UB on WD or accessory budding from the main ureter | Debiec et al.100 | |
| Unknown | |||
| Pax2+/- | Premature branching with incomplete duplication, linked with inactivation of GDNF expression | Brophy et al.101 | |
| Pax2-Cre Lim1Δ/Δ | WD fails to extend caudally; UB is absent or Y-shaped | Pedersen et al.9 | |
| Cc2d2a, Mks1, Cep290, Dync2h1, Tbc1d32, Tmem67 | Duplex kidney as a part of a ciliopathy phenotype | San agustin et al.86 | |
| Sox17Y259N/+ | Duplicated pyeloureteral collecting system | Gimelli et al.76 | |
| Nfia-/- | Partial ureteral duplication | Lu et al.102 | |
| Adamts18-/- | Complete ureteral duplication, increased nephron endowment | Rutledge et al.103 |
As we have seen, the seemingly simple event of ureteric budding is a highly complicated and stringently controlled process that employs both positive and negative feedback loops. As such, the fact that a large number of genes are involved in duplex kidney formation is not surprising, and future analyses are likely to identify many more factors involved in these abnormalities. However, the incomplete penetrance of phenotypes and the multigenic basis of this malformation make the confirmation of mutations as being disease-causing increasingly difficult. Indeed, our own unpublished data suggest that duplex kidney phenotypes can be highly genetic background–dependent, indicating the presence of modifier genes. Moreover, intergenic/regulatory mutations or epigenetic mechanisms that affect gene expression levels rather than protein function are likely to contribute to disease. Finally, we should keep in mind that, despite a large degree of conservation, mice and humans show significant differences on a developmental and molecular level104–106. Findings in knockout mice are therefore only indicative and should not be directly extrapolated to human patients. Future research should also address the disparity in the frequency of duplex kidneys occurring in men and women. In the long run, the integration of a large amount of whole genome sequencing data coupled with a better understanding of how gene regulation is achieved will be required to corroborate the involvement of genomic changes and predict the phenotypic outcome in duplex kidneys.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)