Keywords
Genomics, QC, MPS, Coverage analysis, Clinical analysis
Genomics, QC, MPS, Coverage analysis, Clinical analysis
Compared to extensive serial Sanger sequencing, exome sequencing can be done at a small fraction of the cost per sample (the same order of magnitude as one average-sized gene) and whole exome sequencing (WES) has been more or less established in the clinic for a few years1. Sequencing technologies are continuing to improve at a rapid pace. The Illumina HiSeq X system, and the more recent Illumina Novaseq 6000, reduce the cost of whole genome sequencing (WGS) to slightly more than $1,000 for sequencing a human genome to 30x coverage2. For the first time it is now possible to analyze complete human genomes within reasonable time and cost. This will further increase the pace of implementation of massively parallel sequencing (MPS) in new areas, such as diagnostics of inherited genetic disease. When analyzing the enormous data volume from WES and WGS, it is important to identify underrepresented genomic regions by calculating and tracking coverage quality control metrics. This is particularly true if the sequence data is used in diagnostics, since low or not exposed regions can lead to false positive or false negative results3,4. There are a number of tools that provide basic coverage and overlap annotation functionalities: PicardTools5, BEDTools6, Sambamba7 and GATK8. These tools are excellent for comparing the overlap of two feature sets using single operations. However, they do not offer a solution for coverage analysis with different thresholds and different genomic and biological features. Moreover there are many laboratories around the world that work in a production setting where new samples are sequenced and analyzed every week. There is no present solution for persistent storage of coverage data that makes comparisons of hundreds or thousands of samples possible. This is essential to locate genomic regions that are hard to sequence and where the local sequencing pipeline gives insufficient information.
Furthermore, to our knowledge there are no tools that support dynamic report generation. To address these needs, we have developed Chanjo, a fast and flexible toolkit for seamless coverage analysis of genomic and biological features across multiple samples. Chanjo has been incorporated into the clinical analysis pipeline at Clinical Genomics Science for Life Laboratory and analysed more than 4500 rare disease WES and WGS samples to date. We believe Chanjo to be a vital tool for clinicians and researchers performing MPS analysis.
Chanjo is written in Python (3.2+). It follows UNIX conventions and is built around text streams that can be incorporated into pipelines (Figure 1). Chanjo is distributed via GitHub, installation is simple and robust thanks to extensive tests. Chanjo loads output files from Sambamba depth7 and stores coverage related statistics, e.g. average coverage and completeness in a SQL database. Completeness is defined as the percentage of bases meeting a user-defined coverage threshold for each genomic interval (Figure 2). Chanjo does not aim to analyze the whole genome, but will limit the analyses to predefined genomic regions of interest defined in BED format.
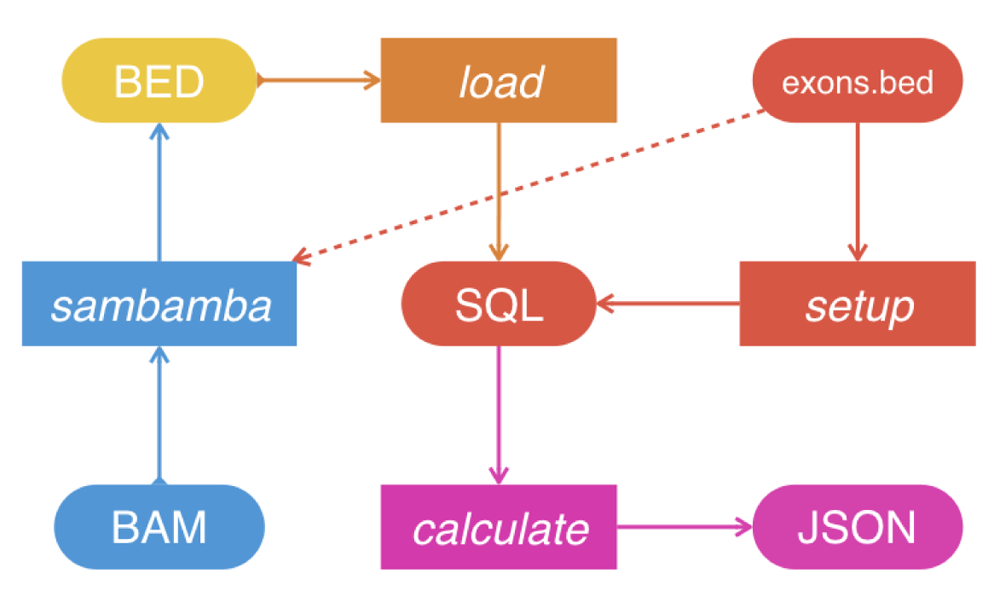
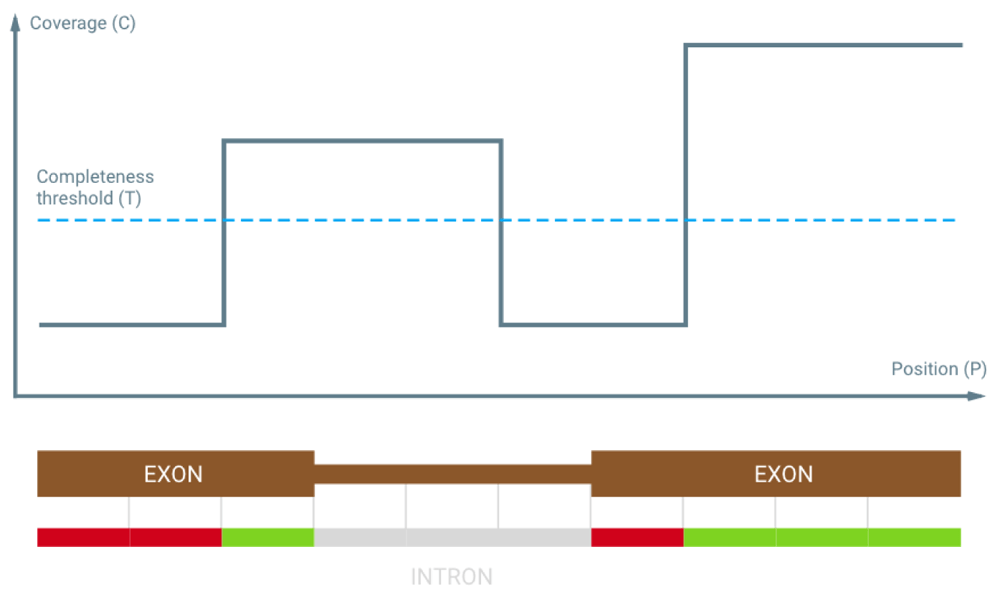
Chanjo supports any genomic intervals as long as they adhere to the BED format with two optional columns for linking exons to transcripts and genes. Hence, it is easy to set-up independent databases using different gene and transcripts definitions, e.g. ccds, refseq or ensembl. When the genomic intervals are defined and added to the database, it is simple to add additional samples.
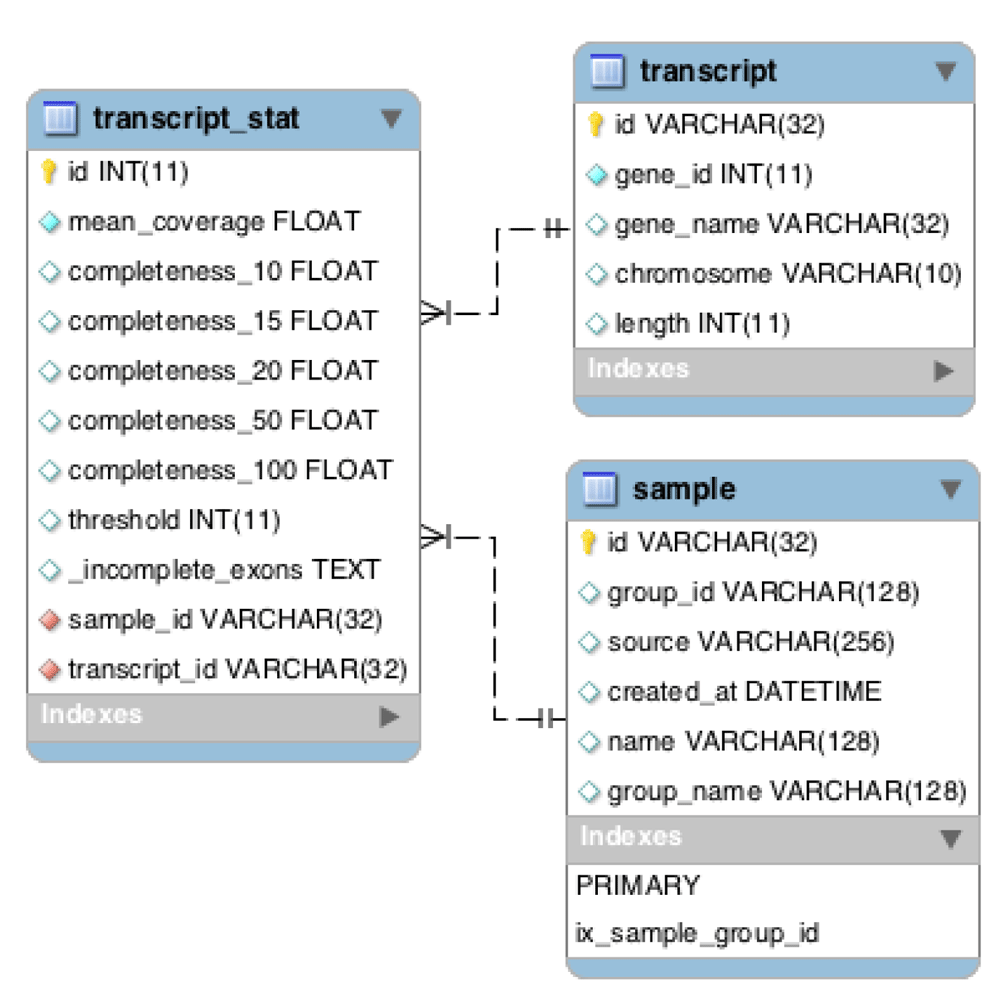
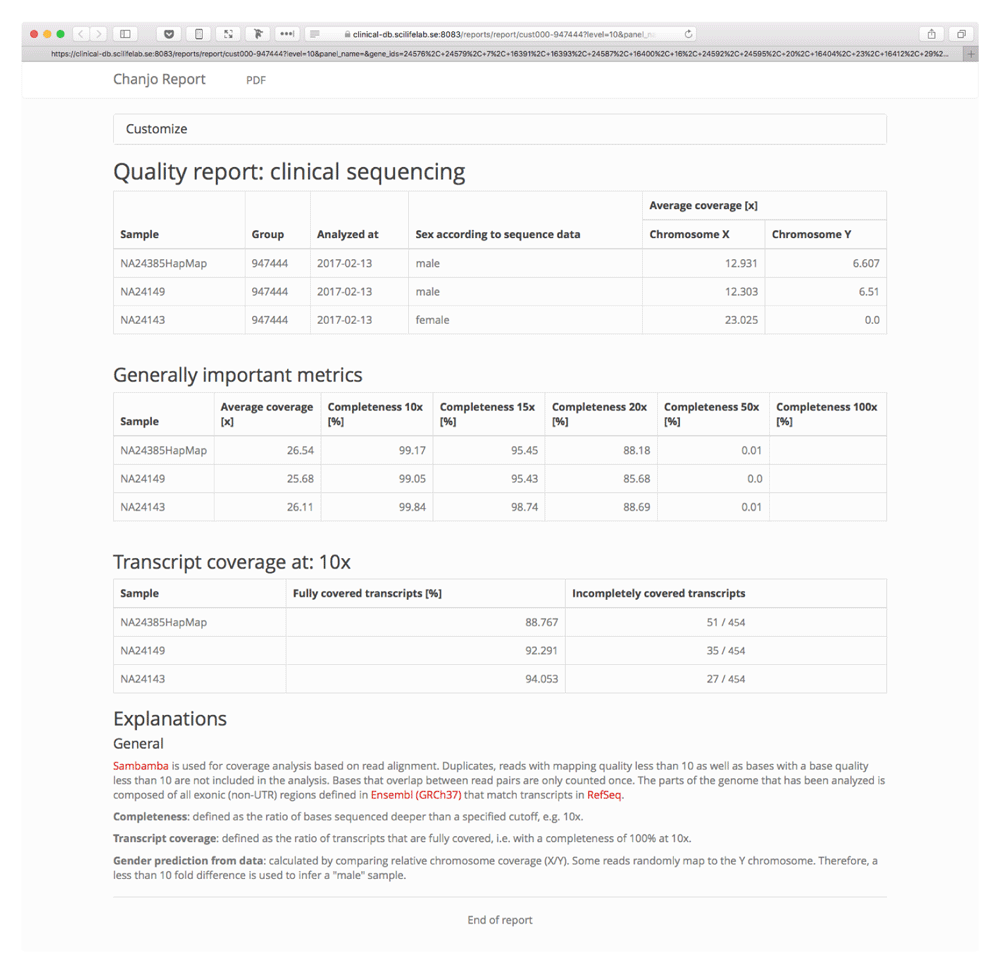
Chanjo uses a predefined database schema where exons are organized into transcripts and genes (Figure 3). Extracting basic coverage metrics such as “average coverage”, “overall completeness”, etc. for different transcripts and genes is easily done through the Python application programing interface (API). Setting up a database with the “init” subcommand only needs to be done once. After the basic structure of the database is in place, the user can add an arbitrary number of samples to the database and include or exclude samples, as preferred from the downstream analysis. The SQL schema has been designed to be a powerful tool on its own for studying coverage. It allows for quick aggregate metrics across multiple samples and can be used as a general coverage API for accompanying tools. One example of such a tool is Chanjo-Report, a clinical-grade coverage report generator for Chanjo output developed to be used in a clinical setting. The report can be tailored to include any number of samples, genes or transcripts (Figure 4). The report can be exported as a PDF and is assembled via a web interface using Chanjo-Report together with a Chanjo database. This works as a powerful bridge between bioinformaticians and professionals that work with analysis who often lack programming skills.
Chanjo requires a installation of Python version 3.2 or above. In a production setting, a mySQL database is preferred, however it is possible to use sqlite, which comes with the python installation. Chanjo has been installed and tested on Linux and Mac OS environments while there should not be any problems to install on Windows. Performance wise, a standard computer will be sufficient to run Chanjo.
It is straight forward to setup a working demo of Chanjo to test how to use the tool:
chanjo init --demo ./ chanjo - demo for file in *. coverage .bed do echo "${ file }" chanjo load --group group1 "${ file }" done chanjo calculate mean -- pretty
The first command initializes a SQL database with tables, it will also use a reduced bed file to link exons, transcripts, and genes according to the definitions in “hgnc.min.bed”. Chanjo uses output generated by “sambamba depth” which includes average coverage and completeness data for each exon defined in “hgnc.min.bed”. The for loop loads data for 3 samples into the database for persistent storage. Finally, the CLI can be used to execute simple queries and output the results in JSON format.
chanjo calculate mean sample_1 { ’metrics ’: { ’ completeness_ 10’: 90.38, ’ completeness_ 20’: 90.92, ’mean_coverage ’: 193.85 }, ’sample_id ’: ’sample_ 1’ }
Chanjo is intended to be used with a central database where samples are continuously added over time. This facilitates aggregate statistics, e.g. trending coverage metrics, poorly covered regions of the genome and comparing gene panel coverage across samples.
In many settings the workflow for a sample starts in the lab where DNA is prepared and sequenced. After that bioinformaticians prepare the output for analysis and hand it over to a researcher or clinician. Chanjo-Report is developed as a tool to present data generated by the bioinformaticians to the end-user. Here one can specify a subset of regions, in many cases a gene panel that is specific for a disease group, and get a well structured report of the fraction of transcripts/genes that are fully covered and which are not. This has proven essential when performing clinical tests with MPS data.
We have developed a novel tool, Chanjo, for continuous and accurate coverage analysis for multiple samples, ideal for WES as well as WGS. We believe Chanjo will be useful for sequencing facilities in general and clinical facilities in particular, where stringent quality control is required. The user only needs to initialize a database once by using a definition of regions and links, e.g. transcripts and genes, in the BED format. Samples are then added by loading data from sambamba depth into the database. Chanjo has been implemented to be easily included in an existing workflow of sequencing analysis. Furthermore, we introduce Chanjo Report to present coverage reports that are easy to generate and to interpret for non-bioinformaticians. This enables, e.g. estimation of accuracy of negative analyses, indicating regions that may require resequencing or investigation using alternative technology.
To our knowledge there are no software freely available today capable of continuous coverage analysis across multiple samples with dynamic report generation like Chanjo.
1. Source code available from: https://github.com/Clinical-Genomics/chanjo
2. Archived source code as at time of publication: http://doi.org/10.5281/zenodo.326649
3. Software license: MIT
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)