Keywords
pneumococcal conjugate vaccine, dosing schedules, expanded programme on immunization
pneumococcal conjugate vaccine, dosing schedules, expanded programme on immunization
We examined three studies comparing immune responses across PCV dosing schedules.
Current dosing schedules are immunologically ineffective and inefficient.
We summarize evidence-based strategies for manipulating PCV dosing schedules.
PCV dosing schedules should leverage immunological data rather than convenience.
The pneumococcal conjugate vaccine (PCV) is arguably the most important and intensely studied vaccine since the Hib conjugate vaccine in the 1980s. It is an extremely safe and effective vaccine, yet complicated both in terms of its immunology and construction1. The technical complexity of its design also makes it very expensive to manufacture. Immunologically, it is not accurate to think of it as one vaccine, but rather 10 or 13 monovalent conjugated vaccines in one syringe, depending on which of the two dominant marketed vaccines one is talking about (PCV10 (Synflorix, Glaxo Smith Klein) or PCV13 (Pfizer), respectively). Logically, such a valuable and costly commodity should be used in the most immunologically astute way possible to yield maximum benefit for its cost. For licensed vaccines, one no longer has the option of changing the vaccine’s composition (choice of carrier protein, adjuvants, antigen content). Instead, the parameters for optimizing the immunogenicity of any licensed vaccine are rooted in details of the dosing schedule2: How early in life can vaccines be started? How many priming doses are optimal? How does the age of infants affect immunogenicity? How far apart should doses be spaced in time? How important is a booster dose? And how soon in life can booster doses be administered?
Until recently, the answers to these pragmatic questions have been elusive due to the near absence of comparative studies exploring the immunogenicity of different dosing regimens. However, in the past few years, a series of pivotal randomized controlled trials focused on dose schedules have been published and shed new light on each of these questions.
In this synthetic analysis, we seek to summarize the key learnings from each of these three seminal investigations, two in high-income settings (The Netherlands and United Kingdom) and one in a low-income setting (Nepal)3–5. It is our contention that these new data suggest that current vaccine schedules used specifically in the US (priming doses at 2, 4, 6 months with a booster at 12 months) and widely in low-income settings through the WHO’s Extended Program of Immunization (EPI) schedule (doses at 6, 10, and 14 weeks, with no booster dose) are immunologically inefficient and either wasteful in terms of administering too many doses of vaccine, or yield suboptimal immune responses due to schedules constructed around expediency rather than based on immunologic data. These learnings present an opportunity to reexamine PCV dosing schedules globally and to align these schedules with immunologic data such that these schedules deliver the greatest long-term cost-effectiveness.
For the purposes of this analysis, our review centers on three recently published randomized controlled trials (Table 1). Given our interest in comparative immunogenicity, we focus on geometric mean concentrations (GMCs) as our outcome of interest. An advantage to GMCs is that there is no ceiling effect in measuring immune responses, as is the case with the proportion of study participants meeting or exceeding some threshold concentration of IgG, and this allows for greater separation of the intrinsic immunogenicity of different dosing strategies. While threshold measures are useful for inferring the proportion of a population that will be seroprotected at a given time, this simplification is distinct from the net magnitude of the immune response. Moreover, GMCs offer some clues as to the likely persistence of protection over time. Moreover, in the case of pneumococci, the commonly used threshold of protection for type specific capsular IgG of 0.35 μg/mL has only been validated for certain serotypes, with the protective relationship assumed to apply to the others6–8. Recent data has made clear that the relationship between IgG GMC and nasopharyngeal carriage varies substantially between serotypes and between high and low incidence settings9–12.
We also note that none of the three studies we reviewed were efficacy studies focused on prevention of clinical disease, so we cannot comment on how these data would translate into morbidity and mortality rates in clinical settings. However, this is an acceptable limitation since field efficacy is already established for PCV, and obviously this is mediated by immunogenicity.
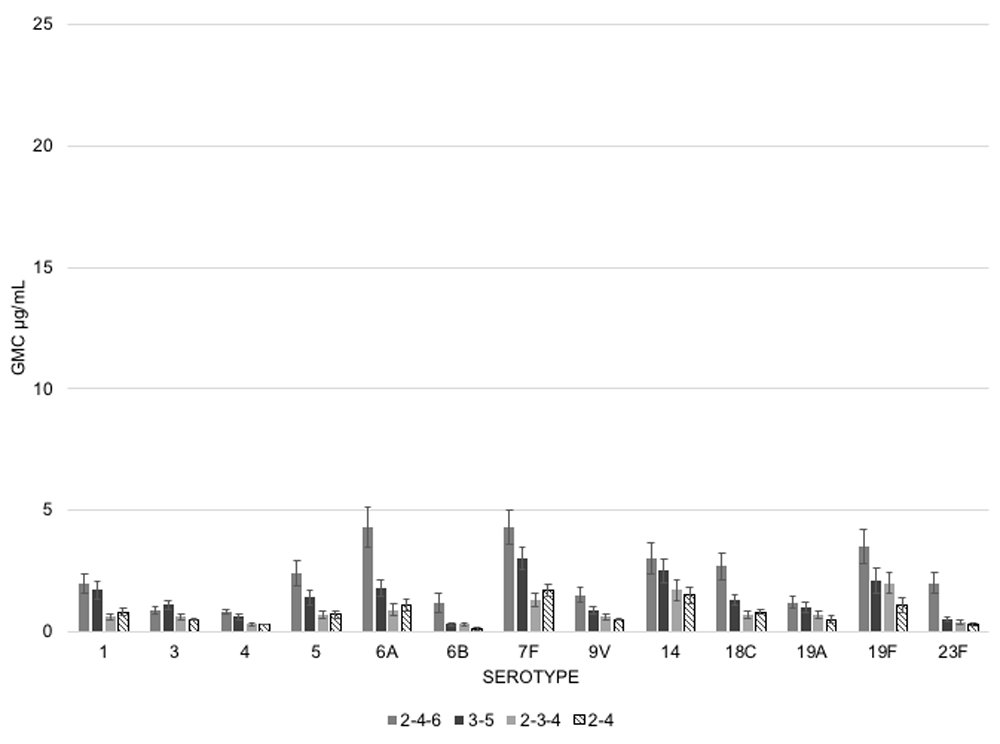
Using a cohort of 400 healthy, term, Dutch infants, Spijkerman et al.3 conducted a large randomized control trial to test the immunogenicity of four different dosing schedules. In this study, infants were randomized (1:1:1:1) to receive three doses of PCV13 either at 2, 4, and 6 months (regimen 1); 2, 3 and 4 months (regimen 2); or to receive just 2 doses of PCV13 at 3 and 5 months (regimen 3) or at 2 and 4 months (regimen 4). Every group received a booster dose at age 11.5 months. To compare differences in immunogenicity across these four schedules, they used observed immunoglobin G (IgG) concentration values (GMC in μg/mL) one month after the primary series and one month after the booster dose.
Upon observation one month post primary series, IgG GMCs for the 2-4-6 schedule were higher for three serotypes than the 2-3-4 and 3-5 schedules. The 3-5 schedule was superior to the 2-4-6 and 2-3-4 schedules for one serotype and the 2-3-4 schedule was higher than the 2-4 schedule for 5 serotypes (Figure 1).

However, these high-level comparisons, in which the statistics sought to measure superiority of one schedule compared to all other schedules, may have significantly understated the observed differences in immunogenicity. When assessing this instead as pairwise comparisons by serotype across schedules, the differences in immune responses to the primary series were more striking. Unsurprisingly, 2-4-6 was statistically superior to 2-4 for 11/13 serotypes. Yet it was surprising that 2-4-6 was superior to 3-5 only for 4/13 serotypes (Table 2). The former is an intuitive finding, but the latter is somewhat surprising and quite interesting: delaying the onset of the primary series (as observed in the 3-5 dosing schedule) seemed to yield better immune responses despite the omission of one of the priming doses (as observed in the 2-4-6 and 2-3-4 schedules). This argues that the timing of doses may be more relevant than the total number of doses in the priming series.
Emphasizing this same point, while also testing the question of dose spacing, the 2-4-6 schedule outcompeted 2-3-4 for 10/13 serotypes, indicating that the increased spacing between priming doses (2 months apart instead of 1) elicited significantly better immune responses. Delayed onset of dosing also appeared immunologically superior given that the 3-5 schedule yielded significantly higher GMCs than the 2-4 schedule for 11/13 serotypes. A surprising and counterintuitive finding was that two doses at 3-5 months yielded superior GMCs than did three doses at 2-3-4 months for 5/13 serotypes. This further demonstrates that the total number of doses given may be less important than the age of the child at the time of vaccine administration.
They also assessed persistence of immune responses following each schedule, testing GMCs again at 8 months (Figure 2) and before the booster dose at 11.5 months (Figure 3). Overall, the 2-4-6 schedule had better persistence at eight months, followed by the 3-5 schedule and both schedules were superior to the 2-4 and 2-3-4. These observations suggest that delaying the onset of doses coupled with the longer intervals between doses yield higher immune responses than dosing schedules where doses are administered earlier in life and closely together. If the goal of dosing schedules is to provide protection early in life, then the 2-4-6 or 3-5 schedules appear to be the most desirable of the four regimens. If we are thinking about this in terms of cost comparisons of these schedules in relation to the total number of doses given, the 3-5 schedule delivers comparable immune responses to the 2-4-6 schedule at a lower price. While the immune responses across all four regimes wane pre-booster, the 2-4-6 and the 3-5 schedules continue to offer the highest residual GMCs.
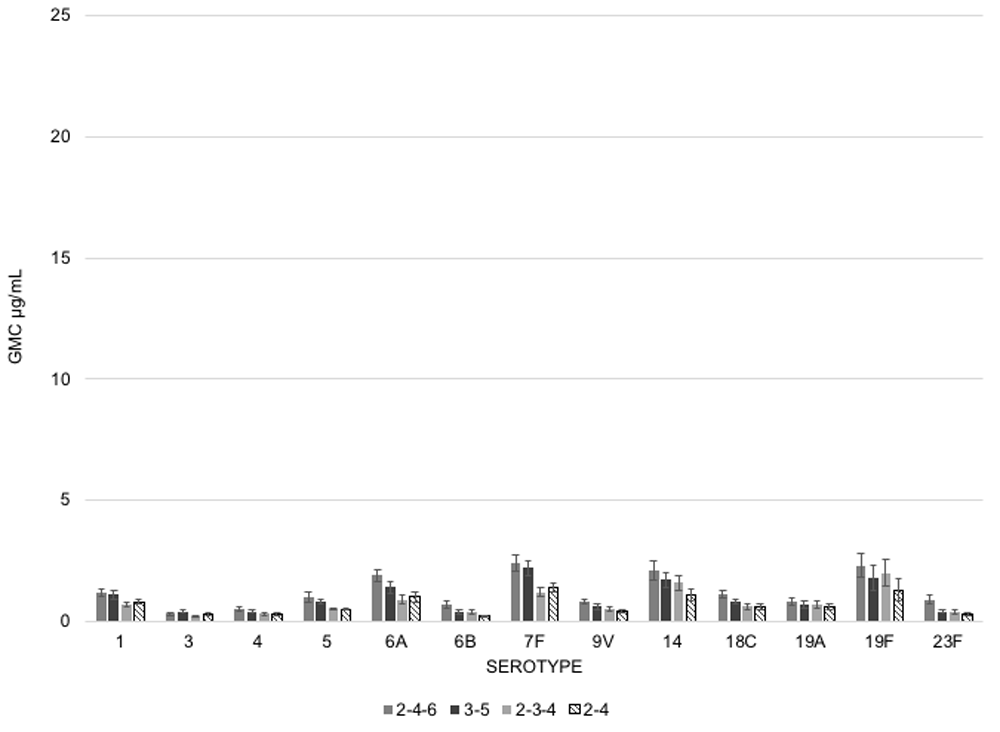
Despite these differences in post primary and pre-boost GMCs, the immune responses following a booster at 11.5 months resulted in significant increases in GMCs, with little variation based on primary schedule (Figure 4). Here, the booster dose acts as the great equalizer between dosing schedules and emphasizes its importance for achieving high immune responses.

In summary, this simple yet elegant experiment yielded a wealth of important and in some cases surprising findings.
First, the absolute number of doses given appeared to be less important than when doses were given. Delaying the onset of PCV administration until later in life (3 months instead of 2 months) may not be a prudent decision for settings that have not reached herd immunity since it leaves infants vulnerable to pneumococcus infection longer. However, in settings with established herd immunity, this herd immunity should provide critical, indirect protection to these infants and allow for flexibility. In these settings, moving the onset of dosing schedules to later in life may not add any undue risk to the infant.
Second, a larger spacing between doses (2 months apart vs. 1 month apart) appeared particularly important, as demonstrated by the fact that 3-5 and 2-4 generally outperformed the 2-3-4- schedule.
Lastly, the booster dose acted as the great equalizer, such that the IgG GMCs were essentially indistinguishable regardless of which priming series had been used.
Also notable was that serotype 3 appeared far less immunogenic than the others, a finding that has been noted elsewhere. Each of these findings has obvious practical implications, as will be discussed subsequently.
Five years later in the UK, Goldblatt et al.4 conducted a similar study to observe how changing the number of priming doses in the PCV dosing schedules would impact serotype-specific IgG GMCs. Unlike the United States, which launched PCV7 using a 2-4-6 priming schedule with a booster at 12 months, the UK instead chose a 2+1 schedule (two priming doses plus a booster in the second year) based on earlier field immunogenicity studies suggesting that this approach would be effective. In the current study, Goldblatt pushed this concept to the next logical point, testing whether a further reduction in priming doses to a 1+1 schedule would be feasible and comparable to the current standard of care. Here, infants were randomized (1:1) to two different dosing schedules, a 2+1 dosing schedule (current standard of care) and an experimental 1+1 dosing schedule. In the 2+1 schedule, infants received two priming doses at 2 and 4 months with a booster dose at 12 months and in the 1+1 schedule, infants received a single priming dose at 3 months and a booster dose at 12 months.
As shown in Figure 5, consistent with expectations, the 2-4 schedule yielded higher GMCs than a single dose at 3 months for 12/13 serotypes. Yet, similar to what was observed in the experiment by Spijkerman et al.3, following the boost at 12 months, GMCs were quite similar between the two schedules, with observed GMC ratios close to 1.0 for most serotypes. Interestingly, while the 2-4 schedule had yielded statistically significantly higher post boost GMC ratios for 4/13 serotypes, a single dose at 3 months yielded significantly superior GMC ratios for 4 of the serotypes. In other words, neither schedule offered a clear advantage over the other. The lack of a clear difference between infants regardless of whether they received one or two priming doses, suggests that a 1+1 dosing schedule could be a less costly but similarly effective alternative to the 2+1 dosing schedule, provided that this is done in a setting where high levels of herd immunity had already been achieved. This finding has obvious practical implications.
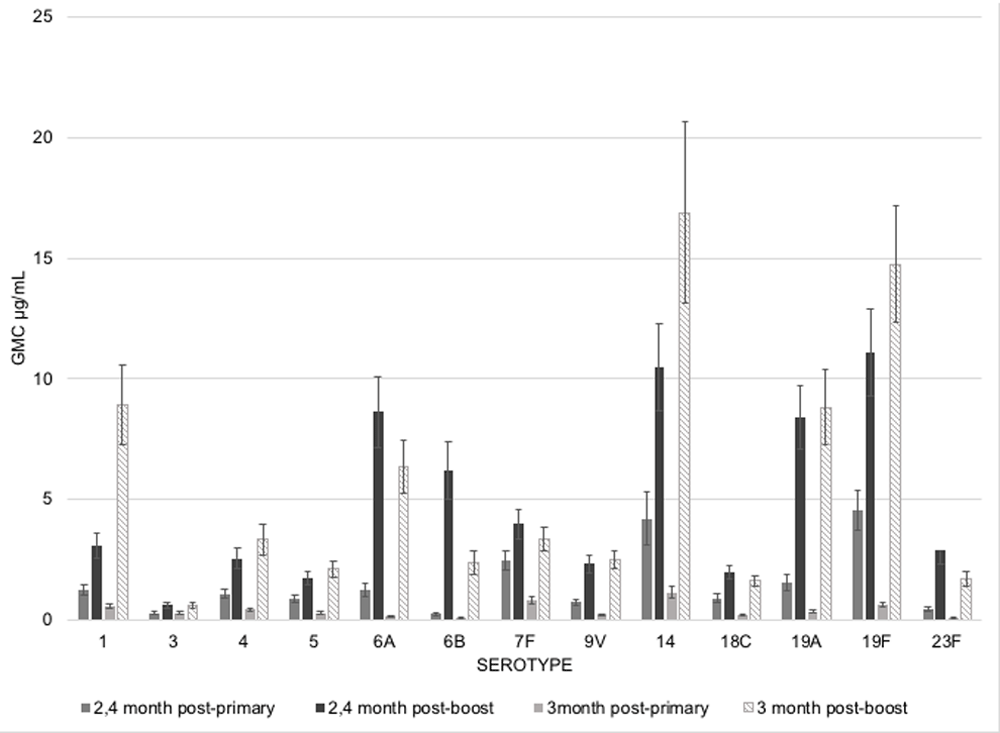
Moreover, when considering the post boost responses in terms of the proportion of infants achieving 0.35 μG of serotype specific IgG (the putative, though somewhat controversial, threshold for protection), 100% of participants were seroprotected against 12/13 serotypes6–8. The exception was again serotype 3, where only 75.9% and 78.8% of the infants randomized to the 2–4 month and 3 month priming schedules, respectively, met or exceeded that threshold.
The studies conducted by Spijkerman et al.3 and Goldblatt et al.4 were both conducted in high-income/low disease burden settings. It is safe to assume that these same issues in terms of dose timing, spacing, and dose number would be at least as important, if not more, in low-income/high disease burden settings. Accordingly, Kandasamy et al.5 conducted a PCV dose schedule study amongst a cohort of Nepalese infants to compare two different dosing schedules, this time using the PCV10 vaccine. Working within the WHO’s EPI schedule of 6-10-14 weeks, infants were randomized (1:1) to receive two priming doses of PCV10 at 6 and 10 weeks (6–10) or at 6 and 14 weeks (6–14), with both groups receiving a booster dose at 9 months of age to complete a 2+1 schedule. The rationale for moving the boost earlier in life to 12 months was to take advantage of the scheduled administration of the measles vaccine at that age, without which a fifth vaccination visit would have to have been created de novo.
To note, the post primary responses for the 6–10 and 6–14 groups were both assessed at 18 weeks. Theoretically, this strategy could penalize the 6-10 regimen to some degree, since it introduces another month for initial antibody concentrations to wane.
With that caveat, as shown in Figure 6, the post primary responses were quite similar between the 6–10 and 6–14 groups, though in general, the 6-14 group GMCs trended higher for most serotypes. These differences were less obvious when reassessed at 9 months, just prior to the boost dose, basically because GMCs had fallen to baseline for most serotypes. However, one month after the 9-month boost, there were robust immune responses to all serogroups (Figure 7). The GMCs were nearly identical for all PCV10 serotypes, irrespective of priming schedule. As seen in the papers by Spijkerman et al.3 and Goldblatt et al.4, once again, the booster dose acted as a grand equalizer, largely erasing the subtle differences following different priming series.
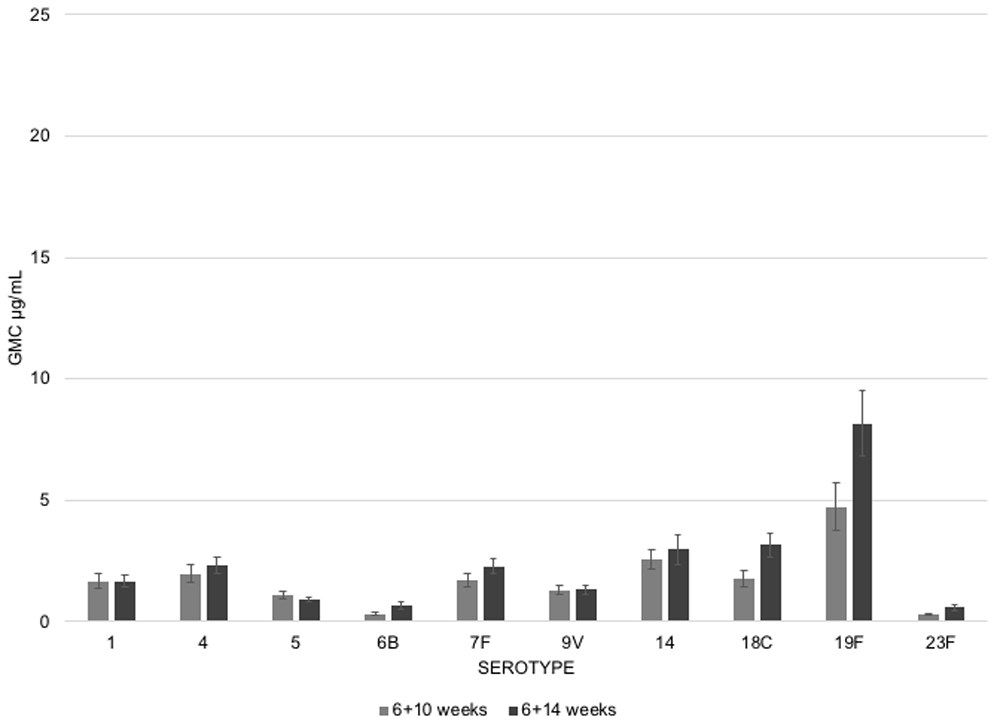

Some exceptions were observed: post-booster GMCs for serotypes 18C and 19F were somewhat higher among infants who received doses at 6-14 than for 6-10. And again, serotype 3 was an immunological dud compared with all other serotypes. Apparently, the comparative underperformance of serotype 3 is not limited to PCV13, but also occurs with other PCV formulations, arguing that the technical construction of the vaccine seems less likely at fault13. Rather, there may be intrinsic limitations in how the immune system engages serotype 3 antigens specifically14.
From this body of evidence, several important messages emerge:
First, that the number of doses in the primary series is less important than at what age doses are given.
Second, in order to maintain optimal levels of antibodies between doses it is important to administer these doses spaced apart by longer intervals.
Third, deferring the start of vaccinations until infants are older, yields higher immune responses. Even a single month’s delay from 2 to 3 months of age yielded markedly higher GMCs for most serotypes.
Fourth, even when infants are dosed on different immunization schedules, the observed differences across these groups largely disappear after the administration of the booster dose. In order to harvest the full benefit of PCV, the booster dose is critically important and simultaneously reassures us that flexibility within the primary dosing schedule is possible. In low disease burden settings, dosing schedules could be reduced or started later in life without compromising protection from infection so long as the booster dose is administered. Any reduction to the number of doses could offer tremendous cost savings15. Being able to align modified vaccine schedules with other essential earlier-life vaccines (e.g. measles vaccine) could also indirectly improve adherence to these schedules.
Fifth, it is not necessary to wait until the second year of life to administer a booster dose. In fact, a booster dose given at 9 months also yielded excellent immunologic responses. While one must be cautious in comparing immune responses between different cohorts (the United Kingdom vs. Nepal, for example), the GMCs seen after a boost at 9 months were remarkably similar to those in the UK infants after a boost at 12 months. This finding has significant practical implications for countries adhering to the EPI schedule where measles vaccine is given at 9 months.
While these messages emerge consistently throughout this body of work, their implications for change are vastly different depending on where you are in the world. For instance, in high-income countries, where herd immunity and disease surveillance programs are well-established, creating a dosing schedule with as few as two doses (one priming dose and a booster dose) may be an effective and cheaper alternative to commonly used dosing schedules. However, in low- and middle-income countries the need for early protection is crucial to child survival and the strategy for adapting the immunization schedule must take this into account.
In conclusion, dosing schedules need not and cannot be designed as “one size fits all”. The lack of scrutiny towards the current PCV schedules has left us with inefficient and ineffective dosing schedules as the status quo. These inefficiencies exist in high- and low-income settings alike. Given that the UK has achieved similar success to the US using a 2+1 vs. 3+1 schedule, and evidence here that a 1+1 schedule is also justifiable, one may conclude that the US schedule is wasteful. Similarly, given the criticality of the booster dose, the EPI schedule of 6, 10 and 14 weeks, with no booster, may be spending more than necessary on doses of vaccine, while using them in a way that immunologically inefficient. Key learnings from this synthesis suggest several strategies for adapting dosing schedules to deliver robust immune responses, including longer dose spacing, fewer doses and the possibility for earlier booster doses. These changes could provide countries with cheaper alternatives to commonly used dosing schedules without compromising protection against pneumococcal disease.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)