Introduction
The brain is the most energy-demanding organ in the body. In humans, the brain comprises 2% of total body mass yet consumes 20% of total oxygen and 25% of glucose1,2, making the brain susceptible to even modest disruptions in energy homeostasis3–5. Indeed, the aging brain, even of healthy aging individuals, is marked by glucose hypometabolism and mitochondrial dysfunction6,7. These metabolic and bioenergetic phenotypes are exaggerated in multiple age-associated neurodegenerative diseases (NDs), including Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS)8–18.
Both preclinical and clinical studies reveal that alteration in brain metabolic status during aging and in ND is accompanied by shifts in energy sources, from glucose metabolism to fatty acid metabolism and ketone bodies19–22. While this strategy serves as an adaptation to sustain ATP production7, it also leads to increased free radical production10,23,24, lipid peroxidation21,22,25,26, oxidative stress27,28, and endoplasmic reticulum (ER) stress17,18. Increased production of damage-associated molecular patterns (DAMPs), such as extracellular ATP29, mitochondrial DNA (mt-DNA), reactive oxygen species (ROS)30, ceramides31, oxidized low-density lipoproteins32, and myelin debris21,33–35, further induces chronic systemic inflammation. Induction of chronic systemic inflammation by metabolic stressors can serve as a missing mechanistic link from metabolic and bioenergetic dysfunction to ND36.
In females, estrogen therapy initiated during the critical windows of peri-menopause to early menopause and surgical menopause has been shown to promote brain glucose metabolism37–46, reduce chronic inflammation47–50, and prevent cognitive decline51–57. Understanding the dismantling process of estrogen-regulated metabolic and immune systems during both chronological and endocrinological aging in the female brain can provide insights into ND prevention, diagnosis, and therapy. In this review, we discuss metabolic changes during pre-menopausal aging, peri-menopausal aging, and post-menopausal aging; their impact on neuroinflammation during each of the chronological and endocrinological transition stages; and the implications for NDs.
Menopause and estrogen regulation of brain metabolism and inflammation
The menopausal transition is characterized by reproductive senescence and loss of ovarian hormones, particularly estrogen, in females. Estrogen regulates the systems of biology required for brain glucose metabolism and mitochondrial function38. Estrogen promotes glucose uptake by both capillary endothelial cells of blood–brain barrier and neurons45,46, increases protein expression, and enhances activity of glycolytic enzymes41,44 and also increases protein expression of electron transport chain (ETC) subunits41–43. In vitro studies using rat embryonic neurons and glial cells also revealed increased maximal respiratory capacity in response to estrogen treatment58. Not only can estrogen promote ATP production in healthy neurons in vitro, it can also preserve ATP production capacity in neurons exposed to Aβ1-4259. In surgically menopausal rodent models, estrogen treatment successfully prevented loss of mitochondrial respiratory capacity40. Beyond promoting mitochondrial bioenergetics in the brain, estrogen can further reduce ROS production60, promote calcium homeostasis, and protect cells from apoptosis61, which collectively will promote mitochondrial function.
Decline in estrogen level during menopause is also associated with an increase in inflammation, marked by the increased expression of pro-inflammatory cytokines—interleukin-8 (IL-8), tumor necrosis factor alpha (TNF-α), IL-6, and interferon gamma (IFN-γ)—in response to T-cell activation47,62,63. Sexual dimorphism, especially with the decline of estrogen, is particularly evident in the immune system64,65. Post-menopausal women have increased CD4/CD8 ratios and T-cell proliferation and activated T cell–mediated autoimmunity66,67. Multiple effects in the periphery are a direct response to the loss of immunosuppressive effects of estrogen. In the brain, estrogen is a master regulator of glucose metabolism, neuronal and glial bioenergetics, and microglial inflammation68. The dysregulation of glucose metabolism in the brain is evident during the menopausal transition and can cause the accumulation of DAMPs, further causing the activation of innate and adaptive immunity to induce chronic low-grade inflammation36.
Hormonal change associated with menopausal transition is a gradual process spanning multiple years, thus allowing adaptation in both metabolic and inflammatory function in the brain. Similarly, chronological aging before and after this endocrinological aging stage is coupled by systematic alterations in metabolic and immune systems. Below, we review these fluctuations in more detail during pre-menopausal, peri-menopausal, and post-menopausal stages.
Chronological aging: prelude to endocrine aging
Aging is associated with a reduction in glucose metabolism and consequent increase in chronic low-grade inflammation69 (Figure 1). Clinical studies revealed that regional cerebral blood flow in mesial frontal cortex is negatively correlated with age in young to mid-life adults70. Meta-analysis in adults between 20 and 50 years of age suggested that the reduction in brain glucose uptake was most likely due to a reduction in brain aerobic glycolysis71. Similar findings were evident in a mouse model of the natural menopausal transition72. In comparison with young female mice, mid-aged females had a significant reduction in brain glucose uptake, which was accompanied by significant down-regulation of neuronal glucose transporter 3 (GLUT3) and reduced glycolytic capacity, as evident by a significant reduction in hexokinase activity72. Decline in glucose metabolic system in the brain was exacerbated in the triple-transgenic AD mouse model72. Furthermore, aging from early to mid-adulthood in female rats was associated with significant down-regulation of both gene and protein expression of insulin-like growth factor 1 (IGF-1) in the hippocampus73, suggesting that early disruption in insulin or IGF-1 signaling may underlie changes in brain glucose metabolism during this stage.
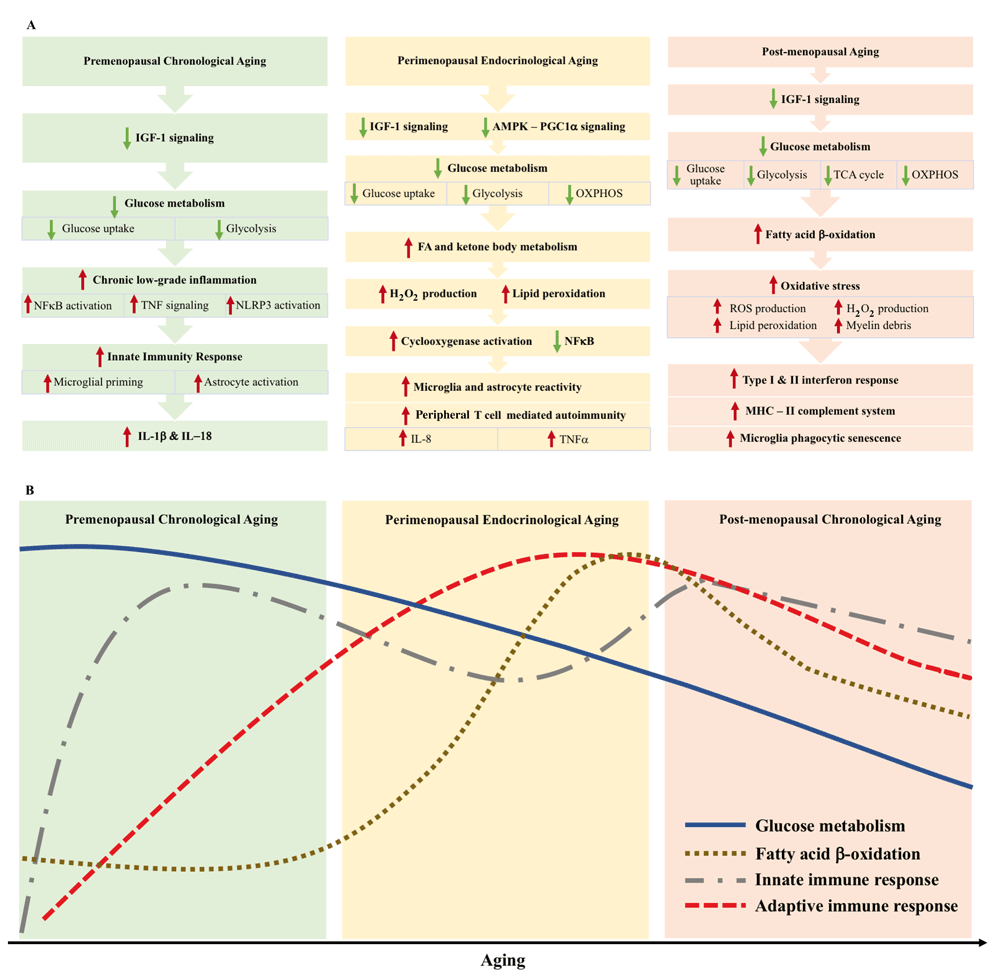
Figure 1. Metabolic and immune signaling during chronological and endocrinological transitions in the mid-life female brain.
(A) Summary of the transition in metabolic and inflammatory female aging in the brain. AMPK-PGC1α, AMP-activated protein kinase–peroxisome proliferator-activated receptor gamma coactivator 1-alpha; FA, fatty acid; H2O2, hydrogen peroxide; IGF-1, insulin-like growth factor 1; IL, interleukin; MHC, major histocompatibility complex; NFκB, nuclear factor kappa B; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle; TNF, tumor necrosis factor. (B) Temporal conceptualization of transitions in glucose metabolism, β oxidation, and innate and adaptive immune response during the course of female brain aging.
Decline in glucose metabolism was specific to the endocrine aging transition as comparable changes were not evident in the reproductively competent animals. No changes in the redox system, including total brain glutathione (GSH) level, GSH peroxidase activity, superoxide dismutase activity, and H2O2 clearance capacity, were observed in reproductively active female rats between early to mid-adulthood74. Similarly, no significant changes were observed in brain synaptic mitochondrial total GSH, lipid peroxides, and cytochrome c oxidase levels in female mice between 10 and 24 weeks of age75. These observations are expected given the relatively steady level of brain and plasma estrogen level during pre-menopausal aging.
Early indicators of disruption in glucose metabolism and IGF-1 signaling during the peri-menopausal phase are associated with increased inflammation through the activation of the inflammatory sensors of aging, nuclear factor-kappa B (NFκB) and TNF76 (Figure 1). In a peri-menopausal animal model (PAM), activation of NFκB pathway and TNF-related genes occurred during the chronological aging phase preceding the peri-menopausal transition. Activation of NFκB can also cause increased expression of Nod-like receptor pyrin domain-3 (NLRP3) inflammasome complex77. The NLRP3 inflammasome complex is susceptible to an aging-related increase in insulin resistance and the onset of glucose hypometabolism during pre-menopausal aging78,79. The NLRP3 inflammasome complex is responsive to triggers such as age-associated DAMPs, including oxidized mt-DNA and extracellular ATP production due to the onset of metabolic dysfunction20,21, which initiate a cascade of chronic low-grade inflammation in the brain80.
The two-step activation of NLRP3 inflammasome, which is an “immuno-metabolic sensor of aging”, leads to the priming of microglial cells81. Secondary triggers such as extracellular ATP and mt-DNA cause the secretion of pro-inflammatory cytokines IL-1β and IL-1882. Interestingly, ketone body β-hydroxybutyrate mitigates the activation of NLRP3 inflammasome complex83. Pre-menopausal aging is also associated with increased expression of complement genes in the hippocampus, where complement C4-A (C4A) acts as an upstream regulator20.
Therefore, alterations in the metabolic profile in the brain can invoke an innate immune response from resident immune cells – microglia and astrocytes (Figure 1). Simultaneous shifts in the metabolic phenotype lead to sustained chronic inflammatory responses, which when coupled with dysregulated steroidal hormone levels can exacerbate inflammation.
Peri-menopause: metabolic-immunological transition
The peri-menopausal transition in females is defined by irregular menstrual cycles and decline in ovarian and brain estrogen production19,84. This endocrinological transition is associated with the early staging that dismantles estrogen regulation of brain bioenergetics (Figure 1). Brain glucose uptake is gradually and significantly reduced during the peri-menopausal transition, especially in brain regions such as temporal lobe, precuneus, and frontal lobe, and is positively correlated with mitochondrial cytochrome oxidase activity7,20,85,86. As reviewed above, pre-menopausal aging is associated with decreased glycolysis but relatively unchanged oxidative phosphorylation, and mechanistic analyses in rat and mouse natural aging models recapitulating human menopausal transition revealed further reduction in glucose uptake as well as significant down-regulation of brain glucose transporters, key enzymes involved in glycolysis, and oxidative phosphorylation during the peri-menopausal transition20,72. Transcriptomic analysis revealed IGF-1 and AMP-activated protein kinase–peroxisome proliferator-activated receptor gamma coactivator 1-alpha (AMPK-PGC1α) signaling pathways as underlying regulators of metabolic changes20. Brain glucose hypometabolism has also been described as a trigger of hot flashes in peri-menopausal females, an exaggerated compensatory neurovascular response to increase blood flow and glucose delivery to the brain87,88.
Estrogen promotes glucose metabolism in the brain, and loss of estrogen during menopausal transition can lead to utilization of auxiliary fuel sources in the brain, especially fatty acids and ketone bodies20–22. In natural menopausal mouse models, this was evident by the activation of cytoplasmic phospholipase A2 (cPLA2) in the brain21, which was accompanied by increased brain mitochondria H2O2 production and lipid peroxide level21,22. Activation of cPLA2 and production of arachidonic acid are linked to increased inflammation through cyclooxygenase activation and increased prostaglandin and leukotriene secretion89.
Linking these metabolic shifts to ovarian hormones was demonstrated in surgically menopausal rats as evidenced by increased brain lipid peroxidase level and decreased superoxide dismutase activity as well as significantly lower serum triglyceride but higher cholesterol, high-density lipids, and low-density lipids90, a profile consistent with increased fatty acid metabolism. Increased mitochondria lipid oxidation may explain the accumulation of ROS during reproductive aging.
A causal link between metabolic dysregulation and consequent change in the inflammatory profile in the brain during peri-menopause has yet to be established. However, evidence suggests that the regulator of inflammation, nuclear factor kappa B (NFκB), is down-regulated in the hippocampus during peri-menopause20. Meanwhile, in the periphery, T cell–mediated autoimmunity is worsened during peri-menopause and is associated with increased prevalence of rheumatoid arthritis, autoimmune hepatitis, and infectious disorders in women47,66,91.
Decline in estrogen level during peri-menopause can also cause increased expression of adhesion molecules that participate in leukocyte transmigration47. Regions such as the subventricular zone in rodent models that closely surround white matter tracts are particularly susceptible to the leukocyte transmigration92,93. Interestingly, autoimmune symptoms of MS, which generally manifests in early adulthood, are worsened during transition from peri-menopause to menopause94–96. Of note, peri-menopause is marked by significant up-regulation of pro-inflammatory cytokines secreted by CD4 T cells: IL-8 and TNF-α62,63. The occurrence of vasomotor symptoms such as hot flashes during peri-menopause has been correlated with increases in pro-inflammatory cytokines IL-8 and TNF-α62. In contrast, circulating estradiol has an inverse relationship with serum IL-8 levels in peri-menopausal women63. Microglial and astrocytic reactivities increase in response to declining estrogen. Surgical ovariectomy in animals caused increased expression of microglial markers CD14, CD11b, and CD45 and phagocytic markers Fcgr1 and Fcgr2b in the hippocampus and cortex97,98. Collectively, the metabolic-immunological transition of peri-menopause is a tipping point in age-related inflammation in recruiting adaptive responses to the brain (Figure 1).
Post-menopausal aging: profiles for risk and resilience ahead
Circulating and brain estrogen levels are at their lowest in post-menopausal females. Human and animal studies revealed that the brain becomes even less efficient in glucose metabolism and more reliant on lipid as its main fuel source7,20–22. This is evident by reduced regional cerebral blood flow99, brain glucose uptake and ketone body uptake100–103, glycolysis and citric acid (tricarboxylic acid cycle, or TCA) cycle enzyme activities22,104,105, mitochondrial oxidative phosphorylation21,22, and increased enzyme activities of fatty acid β oxidation20–22 (Figure 1). Surgical menopausal rodents also exhibit a higher fasting glucose level, greater brain insulin resistance, and impaired IGF-1 signaling26. The hypothesis that the brain can catabolize its own white matter to generate free fatty acid to fuel itself is supported by high brain cytosolic phospholipase A2 activity, especially in the hippocampus, and accumulation of arachidonic acid in post-menopausal mice21. This process causes an accumulation of myelin debris, a sterile inducer of inflammation21,33. Increase in myelin antigenic load thereby causes phagocytic senescence of microglia and can lead to dysregulated glial metabolism and alteration in extracellular matrix, causing an adaptive response from the periphery.
Meanwhile, oxidative stress accumulates in the brain, where reduced GSH level decreases, GSH disulfide (GSSG) level increases26,106, while ROS production such as H2O2 production and lipid peroxidation increases21,22,26, which have been linked to further inflammatory activation of astrocytes and microglia107 (Figure 1). In the absence of the neuroprotective and anti-inflammatory effect of estrogen, ROS production together with accumulated sterile inflammatory triggers leads the female brain into a chronic inflammatory status107. In ovariectomized rodent models, this is evident by increased expression of microglial reactivity markers – major histocompatibility complex class II (MHC II), CD74, CD86, CD68, and the complement system in the hippocampus and cortex97,98. This microglial molecular signature significantly overlaps with the “late-stage neurodegenerative disease” phenotype, which sees exacerbation of IFN response signaling, and overexpression of MHC genes108. Together, these observations indicate that natural aging, particularly the menopausal transition, exhibits a phenotype of microglia that participates in neurodegeneration. It remains to be understood whether this molecular signature is a beneficial compensation or is the tipping point in the course of neurodegeneration.
Implications for neurodegenerative diseases
The peri-menopausal transition is a tipping point for female brain aging7. From the metabolic perspective, the process begins with decline in glucose metabolism7,20,22,71,72,85,104,105 and increase in insulin resistance20,73, followed by a compensatory mechanism to use fatty acids and ketone bodies as an auxiliary fuel source7,20–22. Furthermore, this process is coupled with increased ROS production, oxidative stress, ER stress, and apoptosis10,17,18,21–28, all of which provoke a neuroinflammatory reaction, to form a vicious circle that activates across metabolic crisis, oxidative and cellular stress, and chronic inflammation109.
On the metabolic front, analysis of postmortem AD brains revealed significantly reduced activities of pyruvate dehydrogenase complex, isocitrate dehydrogenase, and α ketoglutarate dehydrogenase complex, whereas activities of succinate dehydrogenase and malate dehydrogenase were increased12. ETC complex IV activity also declined110–112, as supported by reduced gene and protein expression of complex IV subunits113–115. Similarly, patients with PD have reduced resting-state glucose metabolism in the brain, especially in cortical regions and motor networks85,116,117, and mitochondria from cultured PD neurons also demonstrated reduced ETC activities118,119. Patients with MS have axonal degeneration and oligodendrocyte dysfunction120–122, which have also been attributed to mitochondrial bioenergetic deficiency in neurons and oligodendrocytes122–124 and excessive ROS production121,122,125. Patients with ALS have increased energy expenditure126 accompanied by impaired glucose tolerance127, increased insulin resistance128, and hyperlipidemia129.
Decline in neuronal glucose metabolism and mitochondrial function can serve as an initiating factor for chronic inflammation. Microglial stress response as observed during aging and neurodegeneration is seen through the accumulation of DAMPs due to metabolic dysregulation130. Sterile inducers of microglial inflammation set in motion chronic low-grade inflammation, which leads to premature microglial senescence and excessive synaptic pruning. Specifically, single-cell RNA-sequencing (RNA-seq)-based studies on familial AD models and ALS animal models indicated a disease-associated microglia (DAM) phenotype that is different from that of homeostatic microglia131,132. DAM is characterized by up-regulation of TREM2, APOE, TYROBP, ITGAX, and B2M and down-regulation of CX3CR1, P2RY12, and TMEM119 gene expression132. The shared phenotype of this microglial subpopulation between AD, ALS, and normal aging indicates that a microglial subpopulation dedicated to debris clearance and combating neurodegeneration emerges in the brain.
Engagement of the complement system and phagocytosis are fundamental to synaptic pruning during development, yet dysregulation of this system can lead to excessive loss of synapses133. Dysregulation in complement signaling mediated through complement receptor 3 (CR3) has been implicated in a rotenone-induced PD mouse model134. Microglial ablation achieved by blocking colony-stimulating factor 1 receptor (CSF1R) signaling without reducing amyloid-β load in the brain was beneficial in restoring behavioral deficits and synaptic function135,136. While microglial ablation leads to complete loss of microglia (including homeostatic and DAM microglia), regulation of inhibitory checkpoint signals such as CX3CR1 that play a prominent role in DAM expansion could be pivotal to the development of ND therapeutic strategies131. Activation of inflammasome complex such as NLRP3 and NLRC4 also contribute to increased pro-inflammatory cytokine secretion and increased amyloid-β load in AD mouse models131.
Dysregulation of IFN signaling is central to MS pathology and demyelination137. Interferonopathy induced by USP18 down-regulation increases microglial reactivity associated with white matter tracts to cause demyelination138,139. Up-regulation of type I and type II IFN response genes and MHC II has also been documented as a late-stage disease response in animal studies that model progressive neurodegeneration and aging108.
During the peri-menopausal transition, we identified the emergence of a bioenergetic and inflammatory phenotype that is shared between neurodegenerative disorders. Therefore, therapeutically targeting the metabolic and immune profiles that emerge during this transition state could potentially limit the development of at-risk phenotypes for age-related NDs.
Genetic factors for neurodegenerative disease risk
Over the past decades, it became increasingly clear that genetic variances modulate metabolic and inflammatory phenotypes present in at-risk populations and patients with ND. For example, apolipoprotein E (APOE) genotype, particularly APOE4, is a widely recognized risk factor for AD140–149. APOE4 carriers not only have lower brain glucose uptake compared with non-carriers150–155 but also exhibit more severe, more widespread, and more rapid decline in brain glucose hypometabolism150,153,156–158.
Mechanistic studies indicate an association between APOE4 genotype and mitochondrial dysfunction and glucose hypometabolism in the brain150,151,153,156,158–163. APOE4 gene expression in humans was associated with down-regulation of genes involved in mitochondrial oxidative phosphorylation and energy metabolism164,165. In APOE4 knock-in mice, proteomic analysis revealed decreased expression of proteins involved in the TCA cycle, glucose, lipid and amino acid metabolism166.
The impact of metabolic health on cognitive function was investigated in a cohort of healthy post-menopausal females167,168. Outcomes of these analyses indicated that a metabolic profile indicative of risk for metabolic syndrome/type 2 diabetes was associated with significant deficits in verbal memory, executive function, and global cognitive performance, which were more prominent in APOE4 carriers167,168.
Microglia and astrocytes contribute as major cell types in the production of APOE; therefore, the contribution of APOE to innate immune responses can be expected169–173. Up-regulation of APOE expression as part of the DAM phenotype contributes to a microglial phenotype that combats progression of disease phenotype132. Given that the APOE4 allele is considered evolutionarily conserved to protect against viral and bacterial infections, in mouse models of familial AD with the APOE4 risk factor, inflammatory challenges such as lipopolysaccharide (LPS) induced a robust pro-inflammatory reaction172,174,175. APOE4 interferes with microglial clearance function through the down-regulation of insulin-degrading enzymes176,177 and neprilysin178 which further exacerbates accumulation of DAMPs such as amyloid-β and activation of the innate immune response36. These mechanistic findings are indicative of the increased chronic low-grade inflammation clinical profile seen in human APOE4 carriers, who have increased expression of C-reactive protein and reduced latency to the onset of AD179.
On the therapeutic side, APOE4-positive patients with mild-to-moderate AD were less responsive to rosiglitazone, which can improve mitochondrial efficiency and glucose metabolism180,181. Interestingly, APOE4 carriers exhibit a better response to non-steroidal anti-inflammatory treatment169,182. Inflammation burden-specific treatment for APOE4 carriers will be critical for the development of APOE4 targeted AD therapeutics169.
Precision treatment strategy and hormone therapy
Given the impact of genetic variance on phenotypes of aging, metabolism, and inflammatory profiles, a personalized precision medicine approach that takes into consideration differences in genetic background, stage of endocrinological/chronological aging, and timing of treatment should be considered when designing future prevention or intervention strategies to promote healthy brain aging in females.
Understanding how the menopausal metabolomic-immuno-crisis drives risk of NDs in females offers insight into prevention and treatment strategies targeted to each chronological and endocrinological aging stage. Furthermore, identification of the subset of females at higher risk for NDs is pivotal to a precision medicine approach for healthy brain aging. Clinical studies have suggested that the combination of APOE genotype and metabolic phenotype can help identify post-menopausal females at risk for cognitive decline167,168.
The data indicate that, during the transition from peri-menopause to menopause, the metabolic-immune systems are in transition from a brain fueled by glucose metabolism to a brain fueled by auxiliary lipid and fatty acid metabolism that generates ketone bodies. This shift in fuel source is mediated in large part by a parallel and interacting shift from an innate immune phenotype to an activated and pro-inflammatory adaptive immune phenotype.
Three key issues for precision hormone therapy require consideration. The first is the limited time window for efficacy of hormone therapy. The introduction of hormone therapy as a preventive versus a treatment intervention has limited windows of efficacy. Efficacy of hormone therapy is limited to when the system is undergoing a transition from peri-menopause to menopause7,53–56,183–190. Hormone therapy has limited to no efficacy and is not advised in late post-menopause for either natural or surgical menopausal females57,183–188,190,191. Second, therapeutics should target the metabolic and immune systems of biology rather than single components within these complex systems. Third, hormone or other therapeutics should specifically target stage-specific metabolic and immune signaling pathways (Figure 1). Hormone therapies, particularly estrogen and progesterone, are regulators of systems of biology that promote glucose metabolism and repress inflammatory processes, which can address these issues38,40,68,192–194.
These considerations are born out in studies of early menopausal females in which those receiving estrogen replacement therapy had higher brain glucose uptake, regulated insulin signaling, and sustained cognitive function51,53–56,195–197. In animal studies, estrogen immediately following ovariectomy resulted in improved bioenergetic capacity, insulin resistance, increase antioxidants, and reduced lipid peroxidation relative to untreated animals40–44,198,199.
Use of hormone therapy or estrogen replacement therapy can also mitigate menopause-related neuroinflammation. Estrogen mitigates the inflammatory action of sterile and infectious agents on microglia and astrocytes by down-regulating inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) expression, reducing TNF-α, IL-1β, macrophage inflammation protein-2 secretion, and ROS production200. Estrogen mediates its effect through both intracellular estrogen receptors ERα and ERβ, which are abundantly expressed in microglia and astrocytes. Ovariectomizing rodents increases microglial reactivity and changes the morphology to a pro-inflammatory phenotype200. Preventive estrogen treatment before ovariectomy mitigates the development of pro-inflammatory phenotype of microglia by down-regulating complement and microglial reactivity genes98. Peripheral immune cells also respond to hormone therapy through mitigating pro-inflammatory responses seen during menopause and preventing immune senescence by maintaining lymphocytes and monocyte numbers36.
Collectively, the data indicate that hormone therapy initiated early in the menopausal transition results in sustained brain metabolic viability and prevention of age-related chronic low-grade inflammation and subsequent development of adaptive immune responses related to inflammation and autoimmunity.
Conclusions
Herein, we reviewed metabolic and inflammatory profiles that emerge during female chronological and endocrinological brain aging. Furthermore, analysis of data from a broad range of studies and laboratories indicates that metabolic and immune transitions in the brain are linked to act in concert. The pre-menopausal aging phase is characterized by a decline in glycolysis and glucose metabolism and a rise in innate immune responses. Estrogen dysregulation sets the stage for peri-menopause and causes further decline in glucose metabolism and mitochondrial oxidative phosphorylation. Disruption in estrogen regulation causes an increase in T cell–mediated adaptive responses. During the post-menopausal aging phase, to offset the bioenergetic demand of neurons, the shift from utilization of glucose to the utilization of auxiliary fatty acid fuel sources to generate ketone bodies results in myelin breakdown. Accumulation of myelin debris induces a rise in the IFN response and MHC expression. Parallels to metabolic and immune profiles comparable to those of the prodromal phases of AD and MS emerge during pre- to peri- to post-menopause aging transition. Biomarkers of risk for post-menopausal age-associated ND coupled with biomarkers of therapeutic efficacy remain to be integrated with hormone therapy interventions. In the twenty-first century, precision hormone therapy is feasible given the current technologies and knowledge of menopausal brain health.
Faculty Opinions recommendedReferences
- 1.
Bélanger M, Allaman I, Magistretti PJ:
Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation.
Cell Metab.
2011; 14(6): 724–38. PubMed Abstract
| Publisher Full Text
- 2.
Kety SS:
The general metabolism of the brain in vivo. In Metabolism of the Nervous System. (ed Derek Richter), Pergamon.
1957; 221–237. Publisher Full Text
- 3.
Bozek K, Wei Y, Yan Z, et al.:
Exceptional evolutionary divergence of human muscle and brain metabolomes parallels human cognitive and physical uniqueness.
PLoS Biol.
2014; 12(5): e1001871. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 4.
Fu X, Giavalisco P, Liu X, et al.:
Rapid metabolic evolution in human prefrontal cortex.
Proc Natl Acad Sci U S A.
2011; 108(15): 6181–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 5.
Magistretti PJ, Allaman I:
A cellular perspective on brain energy metabolism and functional imaging.
Neuron.
2015; 86(4): 883–901. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 6.
Mosconi L, De Santi S, Li J, et al.:
Hippocampal hypometabolism predicts cognitive decline from normal aging.
Neurobiol Aging.
2008; 29(5): 676–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 7.
Brinton RD, Yao J, Yin F, et al.:
Perimenopause as a neurological transition state.
Nat Rev Endocrinol.
2015; 11(7): 393–405. PubMed Abstract
| Publisher Full Text
- 8.
Wallace DC:
A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine.
Annu Rev Genet.
2005; 39: 359–407. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 9.
Khusnutdinova E, Gilyazova I, Ruiz-Pesini E, et al.:
A mitochondrial etiology of neurodegenerative diseases: evidence from Parkinson's disease.
Ann N Y Acad Sci.
2008; 1147: 1–20. PubMed Abstract
| Publisher Full Text
- 10.
Trimmer PA, Swerdlow RH, Parks JK, et al.:
Abnormal mitochondrial morphology in sporadic Parkinson's and Alzheimer's disease cybrid cell lines.
Exp Neurol.
2000; 162(1): 37–50. PubMed Abstract
| Publisher Full Text
- 11.
Beal MF:
Mitochondria, free radicals, and neurodegeneration.
Curr Opin Neurobiol.
1996; 6(5): 661–6. PubMed Abstract
| Publisher Full Text
- 12.
Bubber P, Haroutunian V, Fisch G, et al.:
Mitochondrial abnormalities in Alzheimer brain: mechanistic implications.
Ann Neurol.
2005; 57(5): 695–703. PubMed Abstract
| Publisher Full Text
- 13.
Swerdlow RH, Khan SM:
A "mitochondrial cascade hypothesis" for sporadic Alzheimer's disease.
Med Hypotheses.
2004; 63(1): 8–20. PubMed Abstract
| Publisher Full Text
- 14.
Coskun P, Wyrembak J, Schriner SE, et al.:
A mitochondrial etiology of Alzheimer and Parkinson disease.
Biochim Biophys Acta.
2012; 1820(5): 553–64. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 15.
Brinton RD:
The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications.
Trends Neurosci.
2008; 31(10): 529–37. PubMed Abstract
| Publisher Full Text
- 16.
Lin MT, Beal MF:
Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases.
Nature.
2006; 443(7113): 787–95. PubMed Abstract
| Publisher Full Text
- 17.
Paillusson S, Stoica R, Gomez-Suaga P, et al.:
There's Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases.
Trends Neurosci.
2016; 39(3): 146–57. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 18.
Burté F, Carelli V, Chinnery PF, et al.:
Disturbed mitochondrial dynamics and neurodegenerative disorders.
Nat Rev Neurol.
2015; 11(1): 11–24. PubMed Abstract
| Publisher Full Text
- 19.
Fukuda M, Mentis MJ, Ma Y, et al.:
Networks mediating the clinical effects of pallidal brain stimulation for Parkinson's disease: a PET study of resting-state glucose metabolism.
Brain.
2001; 124(Pt 8): 1601–9. PubMed Abstract
| Publisher Full Text
- 20.
Yin F, Yao J, Sancheti H, et al.:
The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity.
Neurobiol Aging.
2015; 36(7): 2282–95. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 21.
Klosinski LP, Yao J, Yin F, et al.:
White Matter Lipids as a Ketogenic Fuel Supply in Aging Female Brain: Implications for Alzheimer's Disease.
EBioMedicine.
2015; 2(12): 1888–904. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 22.
Yao J, Hamilton RT, Cadenas E, et al.:
Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence.
Biochim Biophys Acta.
2010; 1800(10): 1121–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 23.
Cassarino DS, Swerdlow RH, Parks JK, et al.:
Cyclosporin A increases resting mitochondrial membrane potential in SY5Y cells and reverses the depressed mitochondrial membrane potential of Alzheimer's disease cybrids.
Biochem Biophys Res Commun.
1998; 248(1): 168–73. PubMed Abstract
| Publisher Full Text
- 24.
Halliwell B:
Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment.
Drugs Aging.
2001; 18(9): 685–716. PubMed Abstract
| Publisher Full Text
- 25.
Papaioannou N, Tooten PC, van Ederen AM, et al.:
Immunohistochemical investigation of the brain of aged dogs. I. Detection of neurofibrillary tangles and of 4-hydroxynonenal protein, an oxidative damage product, in senile plaques.
Amyloid.
2001; 8(1): 11–21. PubMed Abstract
| Publisher Full Text
- 26.
Chételat G, Landeau B, Salmon E, et al.:
Relationships between brain metabolism decrease in normal aging and changes in structural and functional connectivity.
Neuroimage.
2013; 76: 167–77. PubMed Abstract
| Publisher Full Text
- 27.
Richardson JS:
Free radicals in the genesis of Alzheimer's disease.
Ann N Y Acad Sci.
1993; 695: 73–6. PubMed Abstract
| Publisher Full Text
- 28.
Labuschagne CF, Stigter EC, Hendriks MM, et al.:
Quantification of in vivo oxidative damage in Caenorhabditis elegans during aging by endogenous F3-isoprostane measurement.
Aging Cell.
2013; 12(2): 214–23. PubMed Abstract
| Publisher Full Text
- 29.
Cauwels A, Rogge E, Vandendriessche B, et al.:
Extracellular ATP drives systemic inflammation, tissue damage and mortality.
Cell Death Dis.
2014; 5: e1102. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 30.
Chen Y, Zhou Z, Min W:
Mitochondria, Oxidative Stress and Innate Immunity.
Front Physiol.
2018; 9: 1218. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 31.
Maceyka M, Spiegel S:
Sphingolipid metabolites in inflammatory disease.
Nature.
2014; 510(7503): 58–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 32.
Rhoads JP, Major AS:
How Oxidized Low-Density Lipoprotein Activates Inflammatory Responses.
Crit Rev Immunol.
2018; 38(4): 333–42. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 33.
Chen GY, Nuñez G:
Sterile inflammation: sensing and reacting to damage.
Nat Rev Immunol.
2010; 10(12): 826–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 34.
Rock KL, Latz E, Ontiveros F, et al.:
The sterile inflammatory response.
Annu Rev Immunol.
2010; 28: 321–42. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 35.
Ertunc ME, Hotamisligil GS:
Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment.
J Lipid Res.
2016; 57(12): 2099–114. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 36.
Mishra A, Brinton RD:
Inflammation: Bridging Age, Menopause and APOEε4 Genotype to Alzheimer's Disease.
Front Aging Neurosci.
2018; 10: 312. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 37.
Rasgon NL, Silverman D, Siddarth P, et al.:
Estrogen use and brain metabolic change in postmenopausal women.
Neurobiol Aging.
2005; 26(2): 229–35. PubMed Abstract
| Publisher Full Text
- 38.
Brinton RD:
Estrogen regulation of glucose metabolism and mitochondrial function: therapeutic implications for prevention of Alzheimer's disease.
Adv Drug Deliv Rev.
2008; 60(13–14): 1504–11. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 39.
Maki PM, Resnick SM:
Longitudinal effects of estrogen replacement therapy on PET cerebral blood flow and cognition.
Neurobiol Aging.
2000; 21(2): 373–83. PubMed Abstract
| Publisher Full Text
- 40.
Yao J, Irwin R, Chen S, et al.:
Ovarian hormone loss induces bioenergetic deficits and mitochondrial β-amyloid.
Neurobiol Aging.
2012; 33(8): 1507–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 41.
Nilsen J, Irwin RW, Gallaher TK, et al.:
Estradiol in vivo regulation of brain mitochondrial proteome.
J Neurosci.
2007; 27(51): 14069–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 42.
Stirone C, Duckles SP, Krause DN, et al.:
Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels.
Mol Pharmacol.
2005; 68(4): 959–65. PubMed Abstract
| Publisher Full Text
- 43.
Bettini E, Maggi A:
Estrogen induction of cytochrome c oxidase subunit III in rat hippocampus.
J Neurochem.
1992; 58(5): 1923–9. PubMed Abstract
| Publisher Full Text
- 44.
Kostanyan A, Nazaryan K:
Rat brain glycolysis regulation by estradiol-17 beta.
Biochim Biophys Acta.
1992; 1133(3): 301–6. PubMed Abstract
| Publisher Full Text
- 45.
Cheng CM, Cohen M, Wang J, et al.:
Estrogen augments glucose transporter and IGF1 expression in primate cerebral cortex.
FASEB J.
2001; 15(6): 907–15. PubMed Abstract
| Publisher Full Text
- 46.
Shi J, Simpkins JW:
17 beta-Estradiol modulation of glucose transporter 1 expression in blood-brain barrier.
Am J Physiol.
1997; 272(6 Pt 1): E1016–E1022. PubMed Abstract
| Publisher Full Text
- 47.
Straub RH:
The complex role of estrogens in inflammation.
Endocr Rev.
2007; 28(5): 521–74. PubMed Abstract
| Publisher Full Text
- 48.
Monteiro R, Teixeira D, Calhau C:
Estrogen signaling in metabolic inflammation.
Mediators Inflamm.
2014; 2014: 615917. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 49.
Villa A, Rizzi N, Vegeto E, et al.:
Estrogen accelerates the resolution of inflammation in macrophagic cells.
Sci Rep.
2015; 5: 15224. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 50.
Novella S, Heras M, Hermenegildo C, et al.:
Effects of estrogen on vascular inflammation: a matter of timing.
Arterioscler Thromb Vasc Biol.
2012; 32(8): 2035–42. PubMed Abstract
| Publisher Full Text
- 51.
Phillips SM, Sherwin BB:
Effects of estrogen on memory function in surgically menopausal women.
Psychoneuroendocrinology.
1992; 17(5): 485–95. PubMed Abstract
| Publisher Full Text
- 52.
Sherwin BB:
Estrogen and/or androgen replacement therapy and cognitive functioning in surgically menopausal women.
Psychoneuroendocrinology.
1988; 13(4): 345–57. PubMed Abstract
| Publisher Full Text
- 53.
Bagger YZ, Tankó LB, Alexandersen P, et al.:
Early postmenopausal hormone therapy may prevent cognitive impairment later in life.
Menopause.
2005; 12(1): 12–7. PubMed Abstract
| Publisher Full Text
- 54.
Shaywitz SE, Naftolin F, Zelterman D, et al.:
Better oral reading and short-term memory in midlife, postmenopausal women taking estrogen.
Menopause.
2003; 10(5): 420–6. PubMed Abstract
| Publisher Full Text
- 55.
Maki PM:
Hormone therapy and cognitive function: is there a critical period for benefit?
Neuroscience.
2006; 138(3): 1027–30. PubMed Abstract
| Publisher Full Text
- 56.
Maki PM, Zonderman AB, Resnick SM:
Enhanced verbal memory in nondemented elderly women receiving hormone-replacement therapy.
Am J Psychiatry.
2001; 158(2): 227–33. PubMed Abstract
| Publisher Full Text
- 57.
Rocca WA, Grossardt BR, Shuster LT:
Oophorectomy, estrogen, and dementia: a 2014 update.
Mol Cell Endocrinol.
2014; 389(1–2): 7–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 58.
Yao J, Chen S, Cadenas E, et al.:
Estrogen protection against mitochondrial toxin-induced cell death in hippocampal neurons: antagonism by progesterone.
Brain Res.
2011; 1379: 2–10. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 59.
Diaz Brinton R, Chen S, Montoya M, et al.:
The women's health initiative estrogen replacement therapy is neurotrophic and neuroprotective.
Neurobiol Aging.
2000; 21(3): 475–96. PubMed Abstract
| Publisher Full Text
- 60.
Cadenas E:
Mitochondrial free radical production and cell signaling.
Mol Aspects Med.
2004; 25(1–2): 17–26. PubMed Abstract
| Publisher Full Text
- 61.
Nilsen J, Diaz Brinton R:
Mechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expression.
Proc Natl Acad Sci U S A.
2003; 100(5): 2842–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 62.
Huang WY, Hsin IL, Chen DR, et al.:
Circulating interleukin-8 and tumor necrosis factor-α are associated with hot flashes in healthy postmenopausal women.
PLoS One.
2017; 12(8): e0184011. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 63.
Malutan AM, Dan M, Nicolae C, et al.:
Proinflammatory and anti-inflammatory cytokine changes related to menopause.
Prz Menopauzalny.
2014; 13(3): 162–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 64.
Gubbels Bupp MR:
Sex, the aging immune system, and chronic disease.
Cell Immunol.
2015; 294(2): 102–10. PubMed Abstract
| Publisher Full Text
- 65.
Gubbels Bupp MR, Potluri T, Fink AL, et al.:
The Confluence of Sex Hormones and Aging on Immunity.
Front Immunol.
2018; 9: 1269. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 66.
Klein SL, Flanagan KL:
Sex differences in immune responses.
Nat Rev Immunol.
2016; 16(10): 626–38. PubMed Abstract
| Publisher Full Text
- 67.
Yang Y, Kozloski M:
Sex differences in age trajectories of physiological dysregulation: inflammation, metabolic syndrome, and allostatic load.
J Gerontol A Biol Sci Med Sci.
2011; 66(5): 493–500. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 68.
Rettberg JR, Yao J, Brinton RD:
Estrogen: a master regulator of bioenergetic systems in the brain and body.
Front Neuroendocrinol.
2014; 35(1): 8–30. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 69.
Yin F, Sancheti H, Patil I, et al.:
Energy metabolism and inflammation in brain aging and Alzheimer’s disease.
Free Radic Biol Med.
2016; 100: 108–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 70.
Schultz SK, O'Leary DS, Boles Ponto LL, et al.:
Age-related changes in regional cerebral blood flow among young to mid-life adults.
Neuroreport.
1999; 10(12): 2493–6. PubMed Abstract
| Publisher Full Text
- 71.
Goyal MS, Vlassenko AG, Blazey TM, et al.:
Loss of Brain Aerobic Glycolysis in Normal Human Aging.
Cell Metab.
2017; 26(2): 353–360.e3. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 72.
Ding F, Yao J, Rettberg JR, et al.:
Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer's mouse brain: implication for bioenergetic intervention.
PLoS One.
2013; 8(11): e79977. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 73.
Zhao L, Mao Z, Woody SK, et al.:
Sex differences in metabolic aging of the brain: insights into female susceptibility to Alzheimer's disease.
Neurobiol Aging.
2016; 42: 69–79. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 74.
Heemann FM, da Silva AC, Salomon TB, et al.:
Redox changes in the brains of reproductive female rats during aging.
Exp Gerontol.
2017; 87(Pt A): 8–15. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 75.
Martńez M, Ferrándiz ML, De Juan E, et al.:
Age-related changes in glutathione and lipid peroxide content in mouse synaptic mitochondria: relationship to cytochrome c oxidase decline.
Neurosci Lett.
1994; 170(1): 121–4. PubMed Abstract
| Publisher Full Text
- 76.
Maldonado-Ruiz R, Montalvo-Martínez L, Fuentes-Mera L, et al.:
Microglia activation due to obesity programs metabolic failure leading to type two diabetes.
Nutr Diabetes.
2017; 7(3): e254. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 77.
Frank MG, Weber MD, Watkins LR, et al.:
Stress-induced neuroinflammatory priming: A liability factor in the etiology of psychiatric disorders.
Neurobiol Stress.
2016; 4: 62–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 78.
Yang Y, Wang H, Kouadir M, et al.:
Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors.
Cell Death Dis.
2019; 10(2): 128. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 79.
Swanson KV, Deng M, Ting JP:
The NLRP3 inflammasome: molecular activation and regulation to therapeutics.
Nat Rev Immunol.
2019; 19(8): 477–89. PubMed Abstract
| Publisher Full Text
- 80.
Raval AP, Martinez CC, Mejias NH, et al.:
Sexual dimorphism in inflammasome-containing extracellular vesicles and the regulation of innate immunity in the brain of reproductive senescent females.
Neurochem Int.
2019; 127: 29–37. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 81.
Jo EK, Kim JK, Shin DM, et al.:
Molecular mechanisms regulating NLRP3 inflammasome activation.
Cell Mol Immunol.
2016; 13(2): 148–59. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 82.
Hanamsagar R, Torres V, Kielian T:
Inflammasome activation and IL-1β/IL-18 processing are influenced by distinct pathways in microglia.
J Neurochem.
2011; 119(4): 736–48. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 83.
Youm YH, Nguyen KY, Grant RW, et al.:
The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease.
Nat Med.
2015; 21(3): 263–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 84.
Burger HG, Dudley EC, Hopper JL, et al.:
Prospectively measured levels of serum follicle-stimulating hormone, estradiol, and the dimeric inhibins during the menopausal transition in a population-based cohort of women.
J Clin Endocrinol Metab.
1999; 84(11): 4025–30. PubMed Abstract
| Publisher Full Text
- 85.
Mosconi L, Berti V, Quinn C, et al.:
Perimenopause and emergence of an Alzheimer’s bioenergetic phenotype in brain and periphery.
PLoS One.
2017; 12(10): e0185926. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 86.
Mosconi L, Berti V, Quinn C, et al.:
Sex differences in Alzheimer risk: Brain imaging of endocrine vs chronologic aging.
Neurology.
2017; 89(13): 1382–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 87.
Dormire SL:
The potential role of glucose transport changes in hot flash physiology: a hypothesis.
Biol Res Nurs.
2008; 10(3): 241–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 88.
Joffe H, Deckersbach T, Lin NU, et al.:
Metabolic activity in the insular cortex and hypothalamus predicts hot flashes: an FDG-PET study.
J Clin Endocrinol Metab.
2012; 97(9): 3207–15. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 89.
Choi SH, Aid S, Bosetti F:
The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: Implications for translational research.
Trends Pharmacol Sci.
2009; 30(4): 174–81. Publisher Full Text
- 90.
Altunkaynak BZ, Unal D, Altunkaynak ME, et al.:
Effects of diabetes and ovariectomy on rat hippocampus (a biochemical and stereological study).
Gynecol Endocrinol.
2012; 28(3): 228–33. PubMed Abstract
| Publisher Full Text
- 91.
Mohammad I, Starskaia I, Nagy T, et al.:
Estrogen receptor α contributes to T cell-mediated autoimmune inflammation by promoting T cell activation and proliferation.
Sci Signal.
2018; 11(526): pii: eaap9415. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 92.
Delaney C, Campbell M:
The blood brain barrier: Insights from development and ageing.
Tissue Barriers.
2017; 5(4): e1373897. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 93.
Roberts TK, Buckner CM, Berman JW:
Leukocyte transmigration across the blood-brain barrier: perspectives on neuroAIDS.
Front Biosci (Landmark Ed).
2010; 15: 478–536. PubMed Abstract
| Publisher Full Text
- 94.
Bove R, Vaughan T, Chitnis T, et al.:
Women's experiences of menopause in an online MS cohort: A case series.
Mult Scler Relat Disord.
2016; 9: 56–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 95.
Bove R, Healy BC, Secor E, et al.:
Patients report worse MS symptoms after menopause: findings from an online cohort.
Mult Scler Relat Disord.
2015; 4(1): 18–24. PubMed Abstract
| Publisher Full Text
- 96.
Desai MK, Brinton RD:
Autoimmune Disease in Women: Endocrine Transition and Risk Across the Lifespan.
Front Endocrinol (Lausanne).
2019; 10: 265. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 97.
Sárvári M, Hrabovszky E, Kalló I, et al.:
Menopause leads to elevated expression of macrophage-associated genes in the aging frontal cortex: rat and human studies identify strikingly similar changes.
J Neuroinflammation.
2012; 9: 264. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 98.
Sárvári M, Kalló I, Hrabovszky E, et al.:
Ovariectomy and subsequent treatment with estrogen receptor agonists tune the innate immune system of the hippocampus in middle-aged female rats.
PLoS One.
2014; 9(2): e88540. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 99.
Bentourkia M, Bol A, Ivanoiu A, et al.:
Comparison of regional cerebral blood flow and glucose metabolism in the normal brain: effect of aging.
J Neurol Sci.
2000; 181(1–2): 19–28. PubMed Abstract
| Publisher Full Text
- 100.
Nugent S, Tremblay S, Chen KW, et al.:
Brain glucose and acetoacetate metabolism: a comparison of young and older adults.
Neurobiol Aging.
2014; 35(6): 1386–95. PubMed Abstract
| Publisher Full Text
- 101.
Tauber C, Beaufils E, Hommet C, et al.:
Brain [18F]FDDNP binding and glucose metabolism in advanced elderly healthy subjects and Alzheimer's disease patients.
J Alzheimers Dis.
2013; 36(2): 311–20. PubMed Abstract
| Publisher Full Text
- 102.
López-Grueso R, Borrás C, Gambini J, et al.:
[Aging and ovariectomy cause a decrease in brain glucose consumption in vivo in Wistar rats].
Rev Esp Geriatr Gerontol.
2010; 45(3): 136–40. PubMed Abstract
| Publisher Full Text
- 103.
Shen X, Liu H, Hu Z, et al.:
The Relationship between Cerebral Glucose Metabolism and Age: Report of a Large Brain PET Data Set.
PLoS One.
2012; 7(12): e51517. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 104.
Boumezbeur F, Mason GF, de Graaf RA, et al.:
Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy.
J Cereb Blood Flow Metab.
2010; 30(1): 211–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 105.
Ding F, Yao J, Zhao L, et al.:
Ovariectomy Induces a Shift in Fuel Availability and Metabolism in the Hippocampus of the Female Transgenic Model of Familial Alzheimer's.
PLoS One.
2013; 8(3): e59825. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 106.
Zhu Y, Carvey PM, Ling Z:
Age-related changes in glutathione and glutathione-related enzymes in rat brain.
Brain Res.
2006; 1090(1): 35–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 107.
Gonzalez C, Diaz F, Alonso A:
Neuroprotective Effects of Estrogens: Cross-Talk Between Estrogen and Intracellular Insulin Signalling.
Infect Disord Drug Targets.
2008; 8(1): 65–7. PubMed Abstract
| Publisher Full Text
- 108.
Mathys H, Adaikkan C, Gao F, et al.:
Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution.
Cell Rep.
2017; 21(2): 366–80. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 109.
Abais JM, Xia M, Zhang Y, et al.:
Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector?
Antioxid Redox Signal.
2015; 22(13): 1111–29. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 110.
Parker WD Jr, Filley CM, Parks JK Jr:
Cytochrome oxidase deficiency in Alzheimer's disease.
Neurology.
1990; 40(8): 1302–3. PubMed Abstract
| Publisher Full Text
- 111.
Parker WD Jr, Parks J, Filley CM, et al.:
Electron transport chain defects in Alzheimer's disease brain.
Neurology.
1994; 44(6): 1090–6. PubMed Abstract
| Publisher Full Text
- 112.
Maurer I, Zierz S, Möller HJ:
A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients.
Neurobiol Aging.
2000; 21(3): 455–62. PubMed Abstract
| Publisher Full Text
- 113.
Chandrasekaran K, Giordano T, Brady DR, et al.:
Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease.
Brain Res Mol Brain Res.
1994; 24(1–4): 336–40. PubMed Abstract
| Publisher Full Text
- 114.
Aksenov MY, Tucker HM, Nair P, et al.:
The expression of several mitochondrial and nuclear genes encoding the subunits of electron transport chain enzyme complexes, cytochrome c oxidase, and NADH dehydrogenase, in different brain regions in Alzheimer's disease.
Neurochem Res.
1999; 24(6): 767–74. PubMed Abstract
| Publisher Full Text
- 115.
Kish SJ, Mastrogiacomo F, Guttman M, et al.:
Decreased brain protein levels of cytochrome oxidase subunits in Alzheimer's disease and in hereditary spinocerebellar ataxia disorders: a nonspecific change?
J Neurochem.
1999; 72(2): 700–7. PubMed Abstract
| Publisher Full Text
- 116.
Kuhl DE, Metter EJ, Riege WH, et al.:
Patterns of cerebral glucose utilization in Parkinson's disease and Huntington's disease.
Ann Neurol.
1984; 15 Suppl: S119–25. PubMed Abstract
| Publisher Full Text
- 117.
Borghammer P, Chakravarty M, Jonsdottir KY, et al.:
Cortical hypometabolism and hypoperfusion in Parkinson's disease is extensive: Probably even at early disease stages.
Brain Struct Funct.
2010; 214(4): 303–17. PubMed Abstract
| Publisher Full Text
- 118.
Bose A, Beal MF:
Mitochondrial dysfunction in Parkinson's disease.
J Neurochem.
2016; 139 Suppl 1: 216–31. PubMed Abstract
| Publisher Full Text
- 119.
Grünewald A, Rygiel KA, Hepplewhite PD, et al.:
Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons.
Ann Neurol.
2016; 79(3): 366–78. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 120.
DeLuca GC, Ebers GC, Esiri MM:
Axonal loss in multiple sclerosis: A pathological survey of the corticospinal and sensory tracts.
Brain.
2004; 127(Pt 5): 1009–18. PubMed Abstract
| Publisher Full Text
- 121.
Adiele RC, Adiele CA:
Metabolic defects in multiple sclerosis.
Mitochondrion.
2019; 44: 7–14. PubMed Abstract
| Publisher Full Text
- 122.
Patergnani S, Fossati V, Bonora M, et al.:
Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis.
Int Rev Cell Mol Biol.
2017; 328: 49–103. PubMed Abstract
| Publisher Full Text
- 123.
Mahad D, Ziabreva I, Lassmann H, et al.:
Mitochondrial defects in acute multiple sclerosis lesions.
Brain.
2008; 131(Pt 7): 1722–35. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 124.
Dutta R, McDonough J, Yin X, et al.:
Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients.
Ann Neurol.
2006; 59(3): 478–89. PubMed Abstract
| Publisher Full Text
- 125.
Ghafourifar P, Mousavizadeh K, Parihar MS, et al.:
Mitochondria in multiple sclerosis.
Front Biosci.
2008; 13: 3116–26. PubMed Abstract
| Publisher Full Text
- 126.
Desport JC, Preux PM, Magy L, et al.:
Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis.
Am J Clin Nutr.
2001; 74(3): 328–34. PubMed Abstract
| Publisher Full Text
- 127.
Pradat PF, Bruneteau G, Gordon PH, et al.:
Impaired glucose tolerance in patients with amyotrophic lateral sclerosis.
Amyotroph Lateral Scler.
2010; 11(1–2): 166–71. PubMed Abstract
| Publisher Full Text
- 128.
Reyes ET, Perurena OH, Festoff BW, et al.:
Insulin resistance in amyotrophic lateral sclerosis.
J Neurol Sci.
1984; 63(3): 317–24. PubMed Abstract
| Publisher Full Text
- 129.
Dupuis L, Corcia P, Fergani A, et al.:
Dyslipidemia is a protective factor in amyotrophic lateral sclerosis.
Neurology.
2008; 70(13): 1004–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 130.
Song WM, Colonna M:
The identity and function of microglia in neurodegeneration.
Nat Immunol.
2018; 19(10): 1048–58. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 131.
Deczkowska A, Keren-Shaul H, Weiner A, et al.:
Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration.
Cell.
2018; 173(5): 1073–81. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 132.
Keren-Shaul H, Spinrad A, Weiner A, et al.:
A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease.
Cell.
2017; 169(7): 1276–1290.e17. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 133.
Neniskyte U, Gross CT:
Errant gardeners: glial-cell-dependent synaptic pruning and neurodevelopmental disorders.
Nat Rev Neurosci.
2017; 18(11): 658–70. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 134.
Lecours C, Bordeleau M, Cantin L, et al.:
Microglial Implication in Parkinson’s Disease: Loss of Beneficial Physiological Roles or Gain of Inflammatory Functions?
Front Cell Neurosci.
2018; 12: 282. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 135.
Dagher NN, Najafi AR, Kayala KM, et al.:
Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice.
J Neuroinflammation.
2015; 12: 139. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 136.
Beckmann N, Giorgetti E, Neuhaus A, et al.:
Brain region-specific enhancement of remyelination and prevention of demyelination by the CSF1R kinase inhibitor BLZ945.
Acta Neuropathol Commun.
2018; 6(1): 9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 137.
Ottum PA, Arellano G, Reyes LI, et al.:
Opposing Roles of Interferon-Gamma on Cells of the Central Nervous System in Autoimmune Neuroinflammation.
Front Immunol.
2015; 6: 539. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 138.
Meuwissen ME, Schot R, Buta S, et al.:
Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome.
J Exp Med.
2016; 213(7): 1163–74. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 139.
Goldmann T, Zeller N, Raasch J, et al.:
USP18 lack in microglia causes destructive interferonopathy of the mouse brain.
EMBO J.
2015; 34(12): 1612–29. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 140.
Corder EH, Saunders AM, Strittmatter WJ, et al.:
Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families.
Science.
1993; 261(5123): 921–3. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 141.
Poirier J, Davignon J, Bouthillier D, et al.:
Apolipoprotein E polymorphism and Alzheimer's disease.
Lancet.
1993; 342(8873): 697–9. PubMed Abstract
| Publisher Full Text
- 142.
Saunders AM, Strittmatter WJ, Schmechel D, et al.:
Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease.
Neurology.
1993; 43(8): 1467–72. PubMed Abstract
| Publisher Full Text
- 143.
Rebeck GW, Reiter JS, Strickland DK, et al.:
Apolipoprotein E in sporadic Alzheimer's disease: Allelic variation and receptor interactions.
Neuron.
1993; 11(4): 575–80. PubMed Abstract
| Publisher Full Text
- 144.
Carrieri G, Bonafè M, de Luca M, et al.:
Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer's disease.
Hum Genet.
2001; 108(3): 194–8. PubMed Abstract
| Publisher Full Text
- 145.
Maruszak A, Safranow K, Branicki W, et al.:
The impact of mitochondrial and nuclear DNA variants on late-onset Alzheimer's disease risk.
J Alzheimers Dis.
2011; 27(1): 197–210. PubMed Abstract
| Publisher Full Text
- 146.
Edland SD, Tobe VO, Rieder MJ, et al.:
Mitochondrial genetic variants and Alzheimer disease: A case-control study of the T4336C and G5460A variants.
Alzheimer Dis Assoc Disord.
2002; 16(1): 1–7. PubMed Abstract
| Publisher Full Text
- 147.
Coto E, Gómez J, Alonso B, et al.:
Late-onset Alzheimer's disease is associated with mitochondrial DNA 7028C/haplogroup H and D310 poly-C tract heteroplasmy.
Neurogenetics.
2011; 12(4): 345–6. PubMed Abstract
| Publisher Full Text
- 148.
Wang Y, Brinton RD:
Triad of Risk for Late Onset Alzheimer's: Mitochondrial Haplotype, APOE Genotype and Chromosomal Sex.
Front Aging Neurosci.
2016; 8: 232. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 149.
Riedel BC, Thompson PM, Brinton RD:
Age, APOE and sex: Triad of risk of Alzheimer's disease.
J Steroid Biochem Mol Biol.
2016; 160: 134–47. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 150.
Mosconi L, de Santi S, Brys M, et al.:
Hypometabolism and altered cerebrospinal fluid markers in normal apolipoprotein E E4 carriers with subjective memory complaints.
Biol Psychiatry.
2008; 63(6): 609–18. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 151.
Reiman EM, Caselli RJ, Chen K, et al.:
Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer's disease.
Proc Natl Acad Sci U S A.
2001; 98(6): 3334–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 152.
Mosconi L, Mistur R, Switalski R, et al.:
Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease.
Neurology.
2009; 72(6): 513–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 153.
Reiman EM, Chen K, Alexander GE, et al.:
Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia.
Proc Natl Acad Sci U S A.
2004; 101(1): 284–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 154.
Willette AA, Bendlin BB, Starks EJ, et al.:
Association of Insulin Resistance With Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease.
JAMA Neurol.
2015; 72(9): 1013–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 155.
Small GW, Ercoli LM, Silverman DH, et al.:
Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer's disease.
Proc Natl Acad Sci U S A.
2000; 97(11): 6037–42. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 156.
Mosconi L, Sorbi S, Nacmias B, et al.:
Age and ApoE genotype interaction in Alzheimer's disease: an FDG-PET study.
Psychiatry Res.
2004; 130(2): 141–51. PubMed Abstract
| Publisher Full Text
- 157.
Drzezga A, Riemenschneider M, Strassner B, et al.:
Cerebral glucose metabolism in patients with AD and different APOE genotypes.
Neurology.
2005; 64(1): 102–7. PubMed Abstract
| Publisher Full Text
- 158.
Mosconi L, Nacmias B, Sorbi S, et al.:
Brain metabolic decreases related to the dose of the ApoE e4 allele in Alzheimer's disease.
J Neurol Neurosurg Psychiatry.
2004; 75(3): 370–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 159.
Reiman EM, Chen K, Alexander GE, et al.:
Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism.
Proc Natl Acad Sci U S A.
2005; 102(23): 8299–302. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 160.
Mosconi L, Perani D, Sorbi S, et al.:
MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET.
Neurology.
2004; 63(12): 2332–40. PubMed Abstract
| Publisher Full Text
- 161.
Mosconi L, Herholz K, Prohovnik I, et al.:
Metabolic interaction between ApoE genotype and onset age in Alzheimer's disease: implications for brain reserve.
J Neurol Neurosurg Psychiatry.
2005; 76(1): 15–23. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 162.
Valla J, Yaari R, Wolf AB, et al.:
Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE ε4 allele, the major late-onset Alzheimer's susceptibility gene.
J Alzheimers Dis.
2010; 22(1): 307–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 163.
Wolf AB, Caselli RJ, Reiman EM, et al.:
APOE and neuroenergetics: an emerging paradigm in Alzheimer's disease.
Neurobiol Aging.
2013; 34(4): 1007–17. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 164.
Xu PT, Li YJ, Qin XJ, et al.:
Differences in apolipoprotein E3/3 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease.
Neurobiol Dis.
2006; 21(2): 256–75. PubMed Abstract
| Publisher Full Text
- 165.
Xu PT, Li YJ, Qin XJ, et al.:
A SAGE study of apolipoprotein E3/3, E3/4 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease.
Mol Cell Neurosci.
2007; 36(3): 313–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 166.
Shi L, Du X, Zhou H, et al.:
Cumulative effects of the ApoE genotype and gender on the synaptic proteome and oxidative stress in the mouse brain.
Int J Neuropsychopharmacol.
2014; 17(11): 1863–79. PubMed Abstract
| Publisher Full Text
- 167.
Karim R, Koc M, Rettberg JR, et al.:
Apolipoprotein E4 genotype in combination with poor metabolic profile is associated with reduced cognitive performance in healthy postmenopausal women: implications for late onset Alzheimer's disease.
Menopause.
2019; 26(1): 7–15. PubMed Abstract
| Publisher Full Text
- 168.
Rettberg JR, Dang H, Hodis HN, et al.:
Identifying postmenopausal women at risk for cognitive decline within a healthy cohort using a panel of clinical metabolic indicators: potential for detecting an at-Alzheimer's risk metabolic phenotype.
Neurobiol Aging.
2016; 40: 155–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 169.
Safieh M, Korczyn AD, Michaelson DM:
ApoE4: an emerging therapeutic target for Alzheimer's disease.
BMC Med.
2019; 17(1): 64. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 170.
Tzioras M, Davies C, Newman A, et al.:
Invited Review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimer's disease.
Neuropathol Appl Neurobiol.
2019; 45(4): 327–46. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 171.
Guo L, LaDu MJ, Van Eldik LJ:
A dual role for apolipoprotein e in neuroinflammation: anti- and pro-inflammatory activity.
J Mol Neurosci.
2004; 23(3): 205–12. PubMed Abstract
| Publisher Full Text
- 172.
Finch CE:
Evolution in health and medicine Sackler colloquium: Evolution of the human lifespan and diseases of aging: roles of infection, inflammation, and nutrition.
Proc Natl Acad Sci U S A.
2010; 107 Suppl 1: 1718–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 173.
Tenger C, Zhou X:
Apolipoprotein E modulates immune activation by acting on the antigen-presenting cell.
Immunology.
2003; 109(3): 392–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 174.
Tai LM, Ghura S, Koster KP, et al.:
APOE-modulated Aβ-induced neuroinflammation in Alzheimer's disease: current landscape, novel data, and future perspective.
J Neurochem.
2015; 133(4): 465–88. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 175.
Jofre-Monseny L, Loboda A, Wagner AE, et al.:
Effects of apoE genotype on macrophage inflammation and heme oxygenase-1 expression.
Biochem Biophys Res Commun.
2007; 357(1): 319–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 176.
Lee CY, Landreth GE:
The role of microglia in amyloid clearance from the AD brain.
J Neural Transm (Vienna).
2010; 117(8): 949–60. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 177.
Fernandez CG, Hamby ME, McReynolds ML, et al.:
The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer's Disease.
Front Aging Neurosci.
2019; 11: 14. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 178.
Huynh TP, Davis AA, Ulrich JD, et al.:
Apolipoprotein E and Alzheimer's disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins.
J Lipid Res.
2017; 58(5): 824–36. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 179.
Tao Q, Ang TFA, DeCarli C, et al.:
Association of Chronic Low-grade Inflammation With Risk of Alzheimer Disease in ApoE4 Carriers.
JAMA Netw Open.
2018; 1(6): e183597. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 180.
Risner ME, Saunders AM, Altman JF, et al.:
Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease.
Pharmacogenomics J.
2006; 6(4): 246–54. PubMed Abstract
| Publisher Full Text
- 181.
Roses AD, Saunders AM, Huang Y, et al.:
Complex disease-associated pharmacogenetics: Drug efficacy, drug safety, and confirmation of a pathogenetic hypothesis (Alzheimer's disease).
Pharmacogenomics J.
2007; 7(1): 10–28. PubMed Abstract
| Publisher Full Text
- 182.
Hayden KM, Zandi PP, Khachaturian AS, et al.:
Does NSAID use modify cognitive trajectories in the elderly? The Cache County study.
Neurology.
2007; 69(3): 275–82. PubMed Abstract
| Publisher Full Text
- 183.
Espeland MA, Brunner RL, Hogan PE, et al.:
Long-Term Effects of Conjugated Equine Estrogen Therapies on Domain-Specific Cognitive Function: Results from the Women's Health Initiative Study of Cognitive Aging Extension.
J Am Geriatr Soc.
2010; 58(7): 1263–71. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 184.
Coker LH, Espeland MA, Rapp SR, et al.:
Postmenopausal hormone therapy and cognitive outcomes: The Women's Health Initiative Memory Study (WHIMS).
J Steroid Biochem Mol Biol.
2010; 118(4–5): 304–10. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 185.
Rapp SR, Espeland MA, Shumaker SA, et al.:
Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial.
JAMA.
2003; 289(20): 2663–72. PubMed Abstract
| Publisher Full Text
- 186.
Grady D, Yaffe K, Kristof M, et al.:
Effect of postmenopausal hormone therapy on cognitive function: The Heart and Estrogen/progestin Replacement Study.
Am J Med.
2002; 113(7): 543–8. PubMed Abstract
| Publisher Full Text
- 187.
Binder EF, Schechtman KB, Birge SJ, et al.:
Effects of hormone replacement therapy on cognitive performance in elderly women.
Maturitas.
2001; 38(2): 137–46. PubMed Abstract
| Publisher Full Text
- 188.
Pefanco MA, Kenny AM, Kaplan RF, et al.:
The effect of 3-year treatment with 0.25 mg/day of micronized 17beta-estradiol on cognitive function in older postmenopausal women.
J Am Geriatr Soc.
2007; 55(3): 426–31. PubMed Abstract
| Publisher Full Text
- 189.
Zandi PP, Carlson MC, Plassman BL, et al.:
Hormone replacement therapy and incidence of Alzheimer disease in older women: The Cache County Study.
JAMA.
2002; 288(17): 2123–9. PubMed Abstract
| Publisher Full Text
- 190.
Rocca WA, Grossardt BR, Shuster LT:
Oophorectomy, menopause, estrogen treatment, and cognitive aging: Clinical evidence for a window of opportunity.
Brain Res.
2011; 1379: 188–98. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 191.
Rocca WA, Grossardt BR, Shuster LT:
Oophorectomy, menopause, estrogen, and cognitive aging: the timing hypothesis.
Neurodegener Dis.
2010; 7(1–3): 163–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 192.
Yao J, Brinton RD:
Estrogen regulation of mitochondrial bioenergetics: implications for prevention of Alzheimer's disease.
Adv Pharmacol.
2012; 64: 327–71. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 193.
Brenner DE, Kukull WA, Stergachis A, et al.:
Postmenopausal estrogen replacement therapy and the risk of Alzheimer's disease: a population-based case-control study.
Am J Epidemiol.
1994; 140(3): 262–7. PubMed Abstract
| Publisher Full Text
- 194.
Brinton RD:
Estrogen-induced plasticity from cells to circuits: predictions for cognitive function.
Trends Pharmacol Sci.
2009; 30(4): 212–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 195.
Eberling JL, Reed BR, Coleman JE, et al.:
Effect of estrogen on cerebral glucose metabolism in postmenopausal women.
Neurology.
2000; 55(6): 875–7. PubMed Abstract
| Publisher Full Text
- 196.
Jack CR, Wiste HJ, Weigand SD, et al.:
Age, Sex, and APOE ε4 Effects on Memory, Brain Structure, and β-Amyloid Across the Adult Life Span.
JAMA Neurol.
2015; 72(5): 511–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 197.
López-Grueso R, Gambini J, Abdelaziz KM, et al.:
Early, but not late onset estrogen replacement therapy prevents oxidative stress and metabolic alterations caused by ovariectomy.
Antioxid Redox Signal.
2014; 20(2): 236–46. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 198.
Abbas AM, Elsamanoudy AZ:
Effects of 17β-estradiol and antioxidant administration on oxidative stress and insulin resistance in ovariectomized rats.
Can J Physiol Pharmacol.
2011; 89(7): 497–504. PubMed Abstract
| Publisher Full Text
- 199.
Kumar P, Taha A, Kale RK, et al.:
Physiological and biochemical effects of 17β estradiol in aging female rat brain.
Exp Gerontol.
2011; 46(7): 597–605. PubMed Abstract
| Publisher Full Text
- 200.
Villa A, Vegeto E, Poletti A, et al.:
Estrogens, Neuroinflammation, and Neurodegeneration.
Endocr Rev.
2016; 37(4): 372–402. PubMed Abstract
| Publisher Full Text
| Free Full Text
Comments on this article Comments (0)