Keywords
Nanopores, Fungi, Genomics, Mycobiomes
This article is included in the Nanopore Analysis gateway.
Nanopores, Fungi, Genomics, Mycobiomes
Fungal organisms are frequently observed in surgical pathological diagnosis. Recent developments in technology have enabled the identification of fungi using a variety of methods1. These novel methods are based on analyzing fungal genomes or proteomes using fresh tissues2,3. However, these methods are difficult to apply in formalin-fixed and paraffin-embedded (FFPE) tissues used in surgical pathology. Sanger sequencing and immunohistochemistry staining have been used in FFPE tissue, but most medical institutions only identify fungi based on morphological findings4–9. The identification of fungi only by morphological findings can lead to inadequate treatment for patients due to misdiagnosis, which can often result in fatal consequences10. As different antifungal agents are preferentially used at the initial infection stage depending on the fungus, it is necessary to identify the exact fungi present through testing methods in addition to the morphological findings11.
In order to more accurately identify fungi in FFPE tissues, it is essential to use genomic information. DNA markers that could be used to identify fungi include the internal transcribed spacer (ITS) region, small subunit (nrSSU-18S), large subunit (nrLSU-26S or 28S), elongation factor 1-alpha (EF1α), and the largest (RPB1) and second largest (RPB2) subunits of RNA polymerase1,2. Among these markers, ITS could be relatively easily and effectively used for fungal identification, and the database of the ITS regions of fungi is available12–14. In general, sequencing equipment is classified into first- (Sanger sequencing), second- (massively parallel sequencing), and third-generation (real-time and single molecule sequencing) equipment according to key analytical methods15. Third-generation sequencing technology is characterized by direct sequencing of nucleotides without PCR amplification. Oxford Nanopore Technologies (ONT) introduced several sequencing devices using nanopore sequencing technology, which measures the change in current that occurs when a nucleotide sequence passes through a narrow channel16. There is an advantage in identifying each DNA sequence without PCR amplification, and it is expected that the DNA can be effectively detected in spite of DNA degradation during FFPE tissue preparation and storage.
Extracting only fungal DNA from the fungal infection site of FFPE tissue is very difficult. Therefore, we decided to use the mycobiome analysis method. However, no studies have attempted to analyze the mycobiome in FFPE tissue. Therefore, this study was conducted as a pilot study on the development of a method for finding pathogenic fungi using mycobiome analysis in fungal infected FFPE tissues.
The cases were extracted from the pathological examination reports of five medical institutions. We first extracted pathology reports that mentioned the presence of fungi. We then selected typical cases for use as positive controls, cases reported to be difficult to differentiate (briefly, fungi with branched-hyphae, such as Aspergillus species and Mucor species, are often difficult to distinguish morphologically), and cases with additional information related to fungal identification on the pathology report (i.e., identification using culture or sequencing). Through this process, we finally selected 49 cases (case 3-02 and 5–10 were able to confirm the fungal identification results using sequencing). Cases included 21 lung, eight paranasal sinus, seven gastrointestinal tract, five orbit, two skin and mouth, one adrenal gland, one bone, one gum and one liver samples. The average storage period of FFPE tissue was 4.4 years. This study was exempted from obtaining informed consent by the Institutional Review Board of Hallym University Dongtan Sacred Heart Hospital (NON2018-005).
In each case, FFPE tissue was cut into 50 µm (5 µm × 10) sections and collected in a 2.0 ml conical tube. DNA was extracted using the ReliaPrep FFPE gDNA Miniprep System (Catalogue number: A2351, Promega, Madison, Wisconsin, USA) according to the manufacturer's protocol. The DNA extracted from all cases and 10µl DNA was loaded onto 1% agarose gel (Certified Molecular Biology Agarose, #1613101, Bio-Rad Laboratories, Hercules, California, USA, Dyne LoadingSTAR with 100bp DNA Ladder, #A751, Dynebio, Seongnam, Korea, Mupid-One, #AD160, Takara Bio, California, USA) with 100V for 30 minutes running to confirm the presence of DNA. Four universal primers (ITS1, 5'-TCCGTAGGTGAACCTGCGG-3'; ITS2, 5'-GCTGCGTTCTTCATCGATGC-3'; ITS3, 5'-GCATCGATGAAGAACGCAGC-3'; and ITS4, 5'-TCCTCCGCTTATTGATATGC-3') were ordered from Macrogen (Seoul, Korea) with polyacrylamide gel electrophoresis purification17. Multiplex PCR with four primers using SimpliAmp Thermal Cycler (ThermoFisher Scientific, Waltham, Massachusetts, USA) and Taq polymerase (New England Biolabs, M0287S, Ipswich, Massachusetts, USA) was performed as follows: initial denaturation at 95°C for 5 min, followed by 35 cycles of 94°C for 30 s, 52°C for 30 s, and 72°C for 1 min with a final extension step of 72°C for 8 min and cooling at 4°C1. Sequencing was performed using the FLO-MIN106 flow cell (ONT, Oxford Science Park, UK), ligation sequencing kit 1D (SQK-LSK109, ONT) and PCR barcoding expansion 1–12 (EXP-PBC001, ONT) with a MinION (ONT) device in accordance with the manufacturer’s protocol (protocol version: PBGE12_9066_v109_revC_23May2018). Multiplex sequencing was performed for 10–12 cases at a time (83–100 ng DNA per sample). The base calling was carried out according to ONT’s recommendations without parameter changes using MinION Software Release 18.12.6.
We used BLAST+ 2.10.0 on a local PC (Ubuntu 19.10) for analysis18. In brief, we downloaded the ITS database file from NCBI RefSeq Targeted Loci Project (accession number: PRJNA177353, last modified Mar 3, 2020) and then converted it using “makeblastdb” of BLAST+ 2.10.0. This database file contained the full or partial ITS sequence for 11,133 genera of fungi. The RU7 genomic reference sequences of Pneumocystis jirovecii (accession number: GCF_001477535.1), which are not found in this database, were downloaded and compared. The sequence of Actinomyces israelii, a bacterium that looks morphologically similar to that of a filamentous fungus, was downloaded (Actinomyces israelii DSM 43320, whole genome shotgun sequencing project; NZ_JONS00000000.1) and compared. We confirmed the BLASTN search using GRCh38 (accession number: GCF_000001405.26) to determine if the detected DNA corresponds to the human genome.
We converted FASTQ files to FASTA format using “seqtk” on Github. Sequences of each case were searched using BLASTN using the ITS database file without default parameter modification. When the sequence matched in the ITS database, the result with the highest bit score value was selected. We selected five results in order of highest bit score when five or more fungal DNAs were detected in one case.
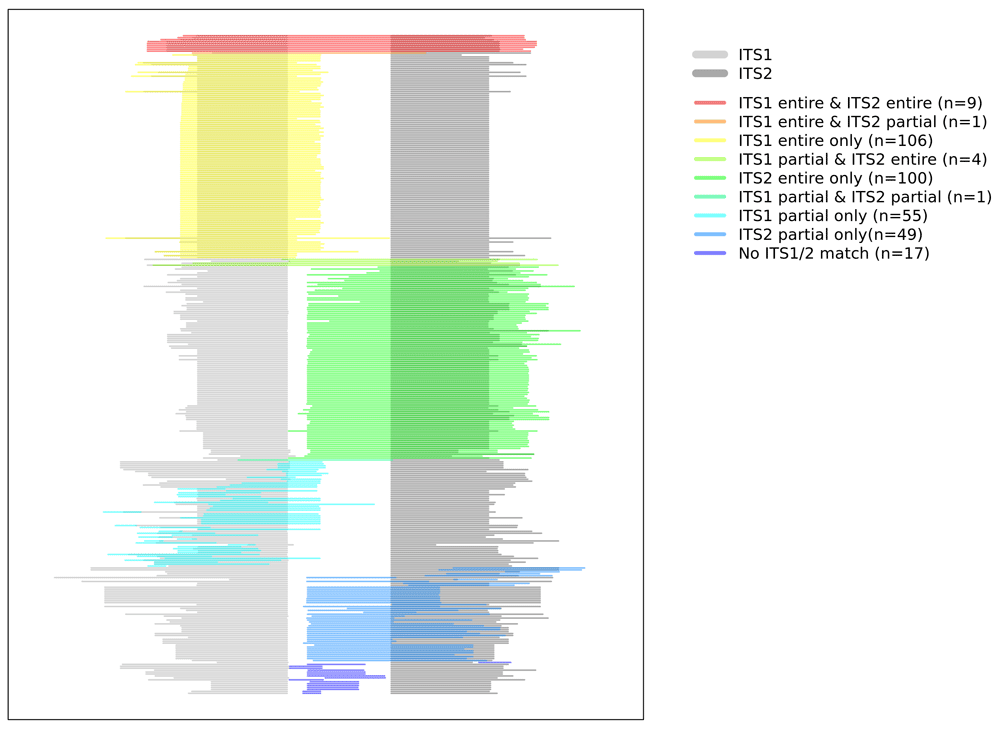
We downloaded the GenBank files of all fungi contained in the database and separated the base sequences of ITS1, 5.8S and ITS2 for each fungus. When fungi were detected, they were classified into three categories (“entire”, “partial”, and “none”) according to the relationship between the detected fungal DNA and ITS1/ITS2. “Entire” means that all ITS1 or ITS2 base information is used for fungal identification, “partial” means that identification is based on a part of ITS1 or ITS2 and its surrounding sequence, and “none” means that base information other than ITS1 or ITS2 is used.
An earlier version of this article can be found on bioRxiv (https://doi.org/10.1101/2020.04.19.045856).
A total of 2,526 DNA sequences were sequenced19. No DNA was sequenced in three cases. Of the remaining 46 cases, the sequenced DNA of 22 cases were not found in the ITS database. We were able to identify 342 fungal sequences in 24 (49.0%, 24/49) cases. The number of DNA sequences identified per case are summarized in Figure 1. The median number of the detected fungal DNA sequences per case was 3 (1Q: 1 and 3Q: 14.25). The clinicopathological information and the number of fungal species detected per case are summarized in Table 1. The remaining cases, in which fungal DNA was not sequenced, are listed in Table 2. Detailed information related to the BLAST program is summarized in Supplementary Table 1 (see Extended data)19. The relationship between the detected fungal DNA and the ITS region is summarized in Table 3 and visualized in Figure 2. Most (215, 62.87%) fungal DNA sequences contained the entire region of ITS1 or ITS2. The remaining 127 fungal DNA sequences were identified as fungi using a partial sequence of ITS1, ITS2, 5.8S, LSU or SSU.
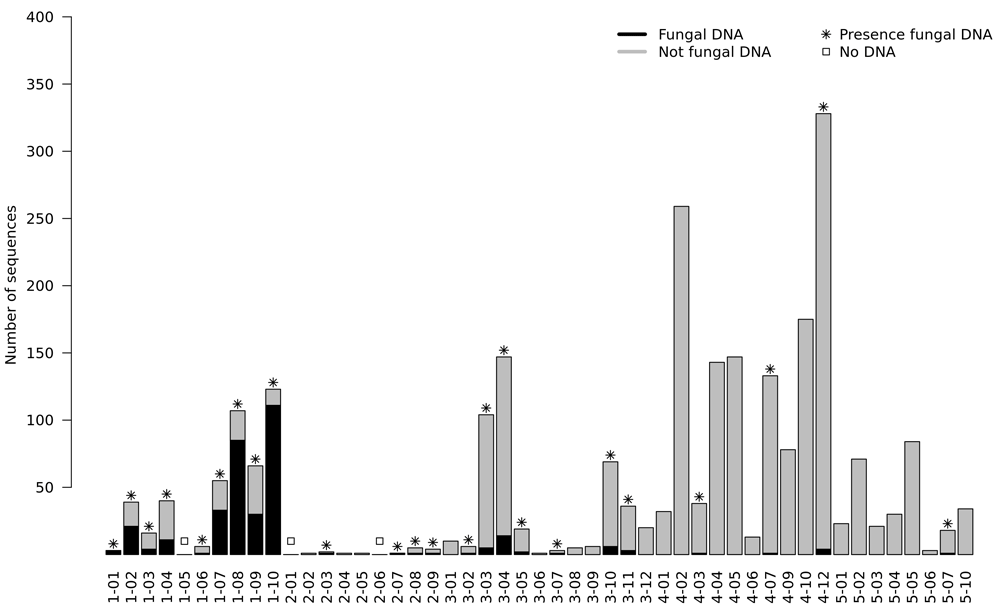
The mean FFPE block storage period of fungal and no fungal cases was 4.2 years and 4.8 years, respectively (p = 0.410). The DNA concentrations measured by NanoDrop after DNA extraction in the groups with and without fungal DNA detection were 71.2 ng/µl and 104.4 ng/µl, respectively, with no statistical significance (p=0.272). The fungal DNA detection rate was not statistically significant (p=0.376)
Representative microscopic images of some cases with inconsistent pathologic diagnosis and mycobiome analysis are summarized in Figure 3 (original images are provided as Underlying data)20. Aspergillus and Candida species were detected by sequencing in case 1-01 (Figure 3A). Compared to the Aspergillus species commonly found in nasal cavities, thinner hyphae were observed, which may have been misleadingly similar to sulfur granules of the pathologically diagnosed Actinomyces. Aspergillus and Acremonium species were detected by sequencing in case 1-03 (Figure 3B). Contrary to case 1-01, it is thought to have been misdiagnosed as Mucor species because its hyphae appeared slightly wider than Aspergillus species. Case 1-08 (Figure 3C) was also misdiagnosed as Actinomyces, in what is thought to be a similar manner to case 1-01. Alternaria, Fusarium and Aspergillus species were detected with numerous sequencing reads. In case 2-08 (Figure 3D), yeast-form fungi were observed in alveolar macrophages using a Gomori methenamine-silver stain and Starmerella cellae was identified by sequencing. Starmerella cellae is a relatively recently identified ovoid to ellipsoidal fungus21. Case 2-09 (Figure 3E) is a fungus found in the pharynx, which shows morphological findings different from those of Candida, Aspergillus, and Mucor. In other words, yeast-form or short branching-type fungal nuclei were found, like in Pneumocystis jiroveci. Cladosporium coloradense was identified by sequencing. Case 3-04 (Figure 3F) is a yeast-form fungus found in subcutaneous tissue and Candida glabrata was identified by numerous sequencing reads.
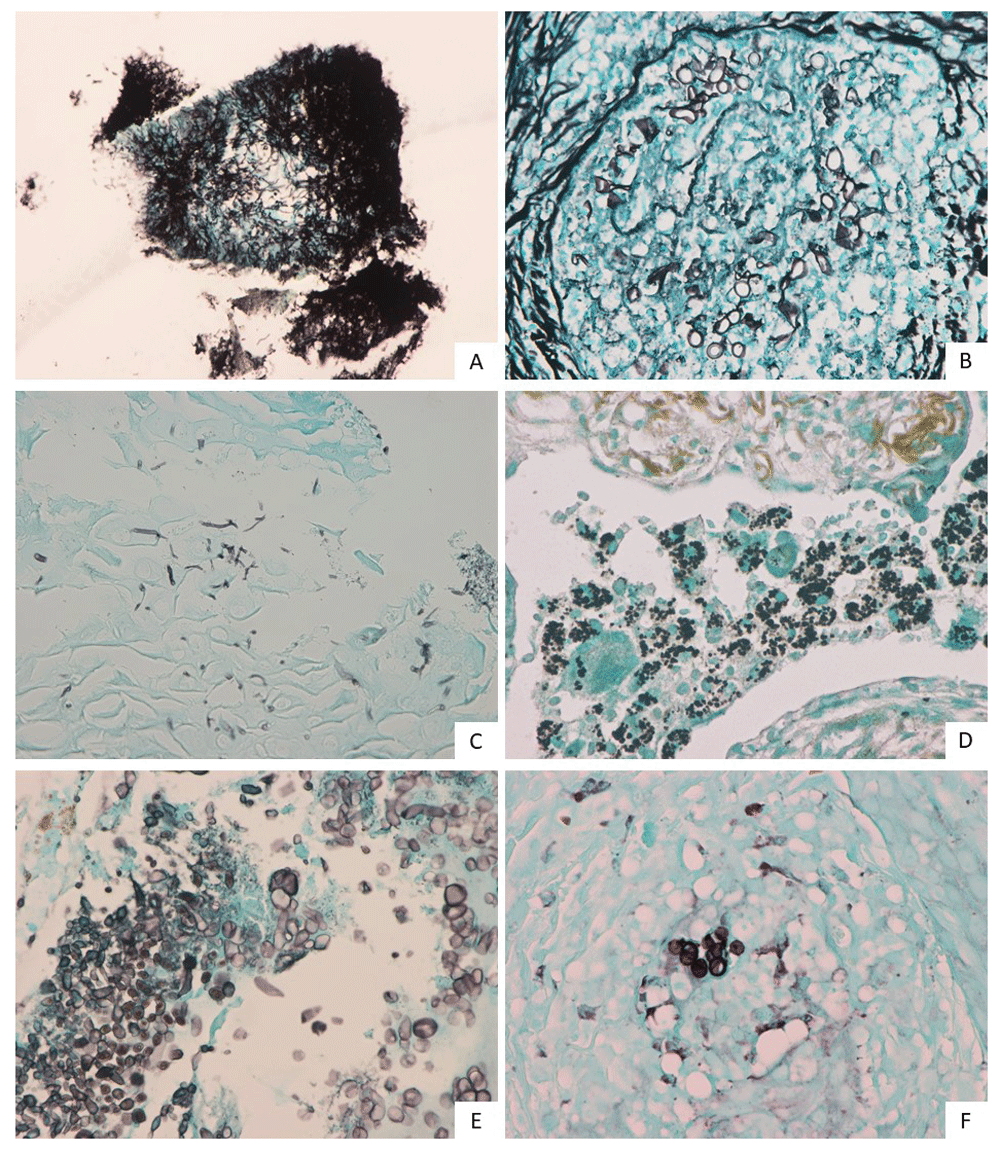
A) case 1-01, B) case 1-03, C) case 1-08, D) case 2-08, E) case 2-09 and F) case 3-04.
All three cases diagnosed with Actinomyces were found to have fungi (cases 1-01, 1-02 and 1-08). Case 3-02 was had a pathological diagnosis of Blastomyces dermatitidis by sequencing analysis (summary of sequencing results: 575 nucleotide sequences of U18364.1 were identical without gap opening and 549 nucleotide sequences of EF592163.1 were identical without gap opening). One strand of fungal DNA was identified from the DNA extracted from the FFPE tissue, and the BLASTN analysis showed the highest probability match was Candida africana with a bit score of 468. In this case, the bit score was 154 based on the separate sequence of Blastomyces dermatitidis. Among three cases of pathologically diagnosed cryptococcosis, one case (case 3-10) was found to have a nucleotide sequence of Cryptococcus neoformans with a bit score of 296. In case 4-12, the nucleotide sequence of Candida africana with a bit score of 438 was suggested, with the possibility of Pneumocystis jiroveci with a bit score of 73.1. Four cases of pathologically unidentifiable fungi (cases 2-03, 2-08, 2-09 and 3-04) were found to most likely be Acremonium acutatum, Starmerella cellae, Cladosporium coloradense, and Candida glabrata, respectively.
We performed mycobiome analysis on fungal infected FFPE tissues using nanopore sequencing. The detected fungal DNA occupied approximately one-third of the ITS1 entire region, the ITS2 entire region, and other regions. The advantages of nanopore sequencing compared with Sanger sequencing are as follows. First, unlike Sanger's method, which requires a lot of DNA, nanopore sequencing can be performed with a small amount of DNA (in theory, even with one strand). Second, the nanopore sequencing method can sequence DNA separately, even if the sample contains a variety of lengths of DNA22. Third, nanopore sequencing equipment (i.e., MinION) can be operated at a lower cost (about $1,000) than Sanger sequencing equipment. This low initial cost is a critical factor in the introduction of equipment in small pathology laboratories. Compared to second-generation sequencing equipment, nanopore sequencing has the advantage of sequencing damaged DNA because there is no PCR amplification process in the sequencing process itself. In this study, about one third of the fungal DNA was not an ITS1 entire match or an ITS2 entire match. Therefore, these fungi can be effectively detected using nanopore sequencing.
It is necessary to find pathogenic fungi among various fungi detected by mycobiome analysis. It may be assumed that dominant fungi are associated with the disease at the site of fungal infection. In cases 1-07 and 1–10, diagnosed as esophageal candidiasis, Candida species comprised 72% and 93% of the fungal DNA, respectively. In such cases, it would be clear to diagnose that the infection is caused by Candida species. In case 1-09, however, only 7% of the fungal DNA was Candida species. Similarly, Rhizopus species were detected in Mucor infections, but as only a small fraction of the fungal DNA. Considering these cases, it is considered desirable to include the process of detecting normal flora in adjacent uninfected sites and selecting pathogenic fungi associated with infection.
We performed multiplex PCR to increase fungal DNA concentration. However, 2,184 non-fungal DNA sequences accounted for 86.46% of the total DNA. Of the 2,184 DNA sequences not identified as fungi, only 382 (17.50%) were identified in the human genome (GRCh38). Because ribosomal DNA is present in all living organisms, including fungi, humans, and bacteria, it may be difficult to amplify only the fungal ITS region. Nevertheless, we were only able to detect fungal DNA in 49.0% of cases. This detection rate is not high enough to be applied in actual clinical situation and needs improvement. There was no difference in the storage period or DNA quality of the FFPE block between cases where fungal DNA was detected and cases that were not detected. We suggest the following about the causes of low fungal DNA detection rates. The first and most important is a low sequencing output. The third-generation sequencing technique is known to have less sequencing output than the second-generation sequencing equipment. Therefore, increasing the purity of the DNA to be sequenced is important for research. Since we used multiplex sequencing, we were forced to reduce the amount of DNA we analyzed per sample. We believe that the lack of sufficient DNA sequencing in each case was the main cause of the failure to detect fungal DNA. The second is the lack of fungal DNA evaluation methods. Unlike human cells, in which cell viability (closely related to DNA quality) could be predicted by evaluation of a haemotoxylin and eosin stained slide, how to evaluate the viability of fungi in a haemotoxylin and eosin stain is not known. Many studies have shown that inflammation and hypoxia are closely related, and hypoxia induced by inflammation caused by fungal infections may result in damage to fungal DNA and consequently affect the sequencing23. Third, the aforementioned ribosomal DNA is present in almost all living things. We believe this could be overcome by using microdissection, which extracts DNA from fungi as much as possible. Fourth, there is a lack of high-quality databases. Even in the database of NCBI RefSeq Targeted Loci Project, there is only one species (murina) in the Pneumocystis genus. In other words, to diagnose Pneumocystis jiroveci, a separate database should be constructed. In addition, when we analyzed the GenBank file, there were 5,602 ITS1 full sequences and 7,552 ITS2 full sequences in 11,133 genera. Therefore, one well-known fungal ITS DB is not sufficient for identification of fungi, and several DBs must be synthesized to analyze the results. Fifth, it is known that tumor biopsy specimens of 50µm thickness can be used to sufficiently perform next-generation sequencing for diagnostic purposes24. Our study was also performed with a 50µm thickness. However, if thicker tissues (e.g., 100µm thick) were used, the fungal detection rate could be increased.
Candida albicans is the most commonly isolated Candida species in the clinical setting. Of the 162 Candida species, 141 Candida africana (87.04%) and eight Candida albicans (4.94%) sequences were detected. These two Candida species are similar in ITS sequences. Comparing the ITS sequences (ITS1+5.8S+ITS2) of Candida africana (NR_138276.1, 447bp) with Candida albicans (NR_125332.1, 446bp), there is 99.192% identity. There is a 1 bp gap in ITS1 in Candida albicans. Two mismatched nucleotides are located in ITS2, where the DNA base "C" in Candida albicans is "T" in Candida africana. It is generally known that nanopore sequencing has a slightly higher error rate than second-generation NGS sequencing25. DNA extracted from FFPE tissue is known to have a higher C:T conversion than DNA extracted from fresh frozen tissue26. Because of these, it is difficult to use our method to precisely identify fungi, especially at the species level.
In summary, we have identified the possibility of finding pathogenic fungi through mycobiome analysis in fungal infected FFPE tissues. However, we have found problems to be solved in further studies, such as increasing sequencing output, increasing fungal DNA concentration, excluding normal flora, and expanding fungal databases. If we develop a method to characterize pathogenic fungi in FFPE tissues in a follow-up study, we think it will help patients to use appropriate antifungal agents.
Figshare: Mycobiome analysis in fungal Infected formalin-fixed and paraffin-embedded tissues for identification of pathogenic fungi: A pilot study. https://doi.org/10.6084/m9.figshare.12616772.v120.
This project contains the following underlying data:
Zenodo: byun1114/mycobiome_1: Mycobiome_anlaysis. https://doi.org/10.5281/zenodo.394271119.
This project contains the following underlying data:
Zenodo: byun1114/mycobiome_1: Mycobiome_anlaysis. https://doi.org/10.5281/zenodo.394271119.
This project contains the following underlying data:
Data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)