Keywords
Fungal Genetics, Fungal Genomics, Genome Evolution, Genetics, Fungi, Genomics, Evolution
Fungal Genetics, Fungal Genomics, Genome Evolution, Genetics, Fungi, Genomics, Evolution
The fungal kingdom is estimated to consist of 2.2 to 3.8 million different species, making it the most diverse kingdom within the eukaryotic domain1. Fungi are currently organized into eight taxonomically distinct phyla2. The most studied and well-characterized fungi belong largely to the Ascomycota and Basidiomycota phyla, which collectively make up the subkingdom Dikarya. The six most basal phyla, also known as early-diverging fungi, are relatively understudied compared to Dikarya fungi and include the Blastocladiomycota, Chytridiomycota, Mucoromycota, Zoopagomycota, Cryptomycota, and Microsporidia phyla2,3.
The diversity within the fungal kingdom is evident from genetic, phenotypic, and ecological perspectives. Fungi display a range of morphologies from macroscopic, multicellular filamentous fungi to environmentally ubiquitous, single-celled yeasts and obligate intracellular pathogens associated with animal hosts. This diversity is further demonstrated by the variety of ecological roles fungi fulfill, the importance of fungi as model organisms in scientific research, and the diversity of applications of fungi in industrial settings. The diverse and important roles of fungi combined with technological advances in high-throughput sequencing, also known as next-generation sequencing, have motivated broad efforts to sequence thousands of fungal genomes4–7. The phenotypic and ecological diversity across the fungal kingdom is reflected, in part, in the diversity observed within fungal genomes.
In this review, we focus on recent advances in understanding the evolution of fungal genomes. Both small- and large-scale variation among fungal genomes contributes to stable or transient genotypic and phenotypic variation as well as speciation. Small-scale genomic changes include changes at the nucleotide and gene level, and often influence genomic microevolution, or the changes of allelic frequencies within a species (Figure 1a). Increased mutation rates and genomic location and context also impact the evolution of fungal genomes8–11. Introgression, meiotic drive elements, and horizontal gene transfer (HGT) from other fungal species as well as cross-kingdom genetic transfer from prokaryotes represent additional examples of small-scale genomic evolution in fungi12–19. Compounding elements of small-scale genomic change can mediate genomic macroevolution, which refers to evolution at and above the species level.
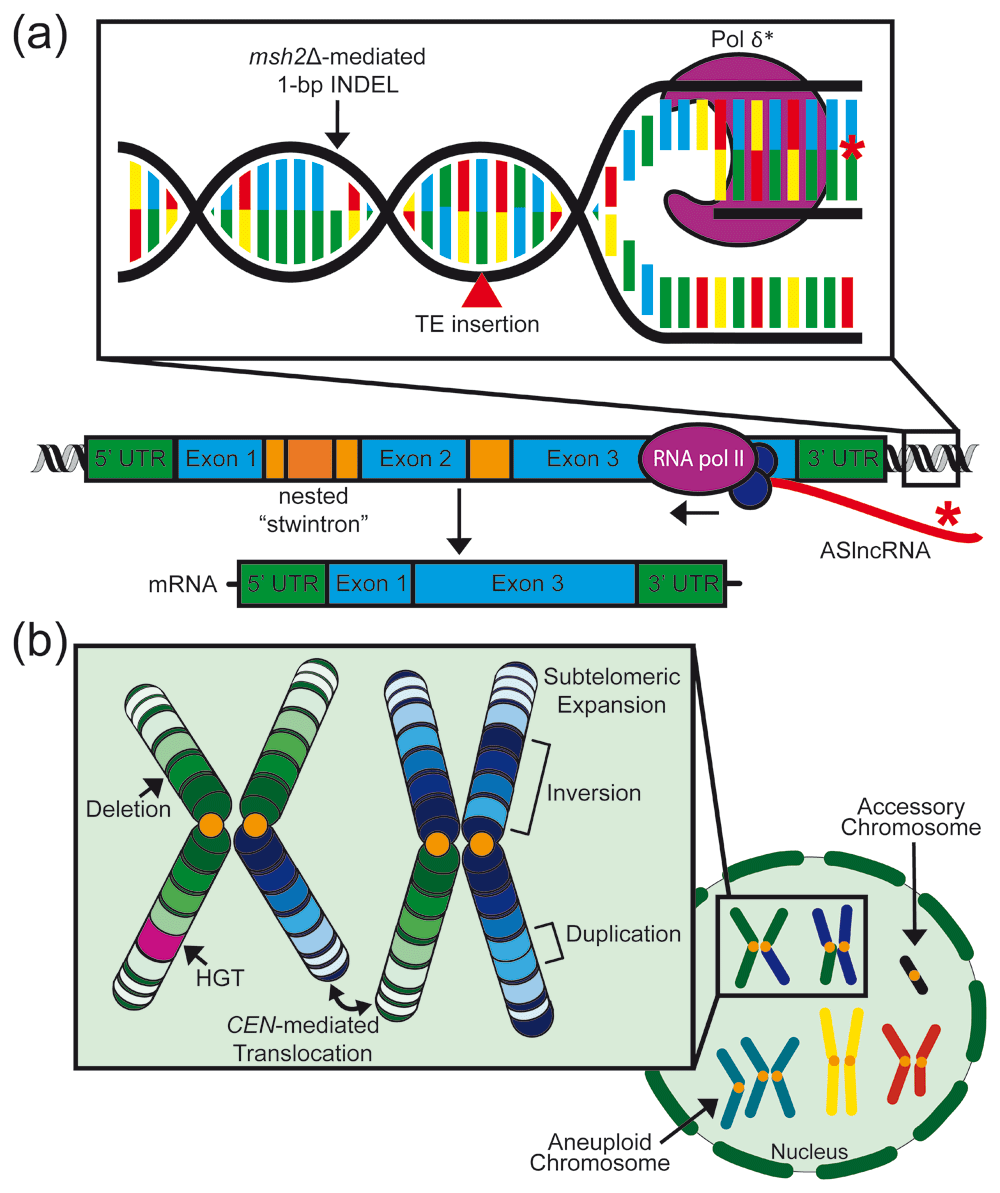
(a) Forces driving genomic evolution in fungi at the nucleotide and gene level Defects in components of the DNA mismatch repair pathway, such as Msh2, contribute to hypermutation. msh2Δ mutants display characteristic 1 base pair (bp) insertions or deletions in homopolymeric nucleotide runs. Transposon insertions and the long terminal repeat footprints they leave behind also influence small-scale genomic evolution. Mutations in DNA polymerase delta subunits (Pol δ*) have been observed in fungal hypermutator isolates and typically generate high rates of transition and transversion mutations. At the gene level, nested introns called “stwintrons” have been shown to mobilize within fungal genomes and can trigger alternative splicing and exon skipping. Long non-coding RNAs (lncRNAs), including antisense lncRNAs (ASlncRNAs), undergo rapid evolution and add one more layer of complexity to the genomic evolution of fungi. INDEL, insertion/deletion polymorphism; Pol, polymerase; TE, transposable element; UTR, untranslated region. (b) Forces driving genomic evolution at the chromosomal and nuclear level Chromosomal structural changes influencing large-scale genome evolution include deletions, inversions, duplications, and centromere (CEN)-mediated translocations. Interspecies horizontal gene transfer (HGT) and the expansion and contraction of subtelomeric regions also contribute to fungal genome evolution. Karyotypic variations including whole-genome duplication, aneuploidy, and the acquisition of accessory chromosomes represent additional instances of large-scale genomic evolution. Orange circles represent centromeres in both the chromosome and nuclear depictions.
Macroevolution of fungal genomes is also influenced by large-scale genomic changes, including structural and organizational changes in individual chromosomes as well as karyotypic variation, which can be attributed to aneuploidy, the acquisition of accessory chromosomes, and polyploidy20–24 (Figure 1b).
Advances in sequencing technologies have allowed robust and rapid characterization of these small- and large-scale genomic changes at a more accurate level than previously possible and therefore have provided novel insights into fungal genomic evolution25–28. These insights contribute to our understanding of fungal genomic evolution and the impacts this evolution can impart on gene function, mating, fitness, and speciation29–31. Genomic evolutionary mechanisms and dynamics in fungi can provide insight into the evolutionary histories and trajectories of species throughout the eukaryotic domain.
Single nucleotide changes, transposition of genetic elements, and expansion and contraction of repetitive sequences influence genome evolution at a small scale. Relatively small-scale genomic changes also include the transfer of genetic information between and among populations. Here we consider advances in understanding how increased mutation rates, fast-evolving genomic regions and elements, and allelic transmission influence the evolution of fungal genomes.
Mutation rates estimate the number of genetic changes occurring within a cell per generation. Isolates with significantly higher mutation rates than those of closely related isolates or laboratory reference strains of the same species are said to display a hypermutator phenotype. Increased mutation rates have been extensively characterized in the yeast Saccharomyces cerevisiae and have provided the foundation for identifying genetic mechanisms underlying human diseases, including a form of hereditary cancer known as Lynch syndrome32–36. Hypermutation in fungi has been shown to promote rapid adaptation to novel environmental conditions, to influence overall fitness and genomic structural changes, and to be subsequently tempered by the emergence of anti-mutator suppressor alleles, among other consequences8,9,37,38.
Recently, naturally occurring fungal hypermutator strains have been identified and characterized. Clinically and environmentally isolated strains of the human fungal pathogens Cryptococcus deuterogattii, Cryptococcus neoformans, and Candida glabrata have been found to harbor loss-of-function mutations in DNA mismatch repair components and thus display hypermutator phenotypes9,39–43. Defects in the mismatch repair protein Msh2 are especially prevalent and typically produce single base pair insertions or deletions in homopolymeric nucleotide runs9,44. These isolates display higher rates of genomic evolution at the nucleotide level and have the ability to more rapidly adapt to novel stressors in vitro, including the ability to develop de novo resistance to antifungal drugs in vitro9,39–42,45. Incompatibilities in the mismatch repair components Mlh1 and Pms1 have also been reported to contribute to hypermutation in S. cerevisiae laboratory strains46,47. Studies have identified several S. cerevisiae strains isolated from humans that encode these same MLH1-PMS1 incompatible alleles48. Although the S. cerevisiae diploids with incompatible alleles isolated from humans were not hypermutators themselves, their meiotic progeny displayed a wide range of mutation rates and included hypermutators, thus providing an opportunity to produce progeny with variable fitness under stressful conditions47,48. Another mechanism of hypermutation caused by a mutation in a DNA polymerase delta subunit was also described in C. neoformans; this mechanism of hypermutation resulted in genome-wide increases in transition and transversion mutations but reduced viability and virulence49,50.
Mobilization of transposable elements throughout the genome can mediate hypermutation at a larger scale than the single-nucleotide polymorphism changes in mismatch repair and DNA polymerase mutants. Within the past year, two studies have reported instances of hypermutation via mobilization of transposons under stress conditions, illustrating that host infection can trigger transposition in the human pathogen Cryptococcus deneoformans and in the plant pathogen Zymoseptoria tritici51,52. Another study has also characterized the evolutionary dynamics and genomic impacts of similar bursts of transposon expansion within the genomes of Microbotryum species53. These instances of hypermutation provide important examples of how elevated genome-wide mutation rates influence rapid genomic microevolution, pathogenesis, and the ability of fungi to rapidly adapt to novel environments.
Despite the fitness benefits of hypermutation in stressful or changing conditions, spontaneous mutations can be largely deleterious in normal conditions, and thus hypermutation might be beneficial only from a short-term evolutionary perspective but disadvantageous in the long term54. All organisms, including fungi, must therefore strike a balance between high mutation rates that provide variation for natural selection to act upon and high genomic integrity that allows for long-term survival. Research has shown that, in contrast to hypermutation, some fungal species, particularly those with relatively long lifespans, can have ultra-low mutation rates, such as those observed in the fairy-ring mushroom species Marasmius oreades and the pathogenic forest fungus Armillaria gallica55,56. On the other hand, some fungi have been found to persist in the long term with high mutation rates and compromised genome integrity. For example, several novel Hanseniaspora species have recently been identified and contributed to the characterization of two Hanseniaspora lineages with different rates of evolution: a fast-evolving lineage and a slow-evolving lineage57. While species in both lineages have lost a number of genes involved in regulating the cell cycle and maintaining genomic integrity, the fast-evolving lineage has undergone rapid genomic changes and represents a group of hypermutator species. These species provide a perspective on the long-term evolutionary consequences of sustained elevated mutation rates and could provide answers to critical questions in the field57.
Genomic evolution can occur at different rates that are dependent on genomic context. Rapid evolution within a genome is driven by the adaptive potential of alleles and relaxed selective constraints as well as the propensity of regions to undergo change. Fungal genomic regions that typically experience faster evolution than other loci include introns, intergenic sequence, centromeres, and subtelomeric regions58–65.
The evolution of introns throughout fungal lineages is dictated by retention of ancestral introns, frequent intron loss events, and relatively few instances of intron gain58,59,66,67. Due to the large variation in intron content across fungal genomes, introns are considered to be one of the more rapidly evolving genomic elements in fungi58,59,66. Spliceosomal twin introns or “stwintrons” were recently identified in several orders of ascomycete species68,69. These nested introns display intriguing trends of gain and loss across fungi and either are removed by complex, multi-step splicing events or trigger alternative splicing and exon skipping. The evolutionary dynamics of these newly identified stwintrons may provide insight into the evolution of intron gain and loss, thus further informing the known evolutionary trajectory of fungal genomes.
The breadth and depth of RNA sequencing in fungi has revealed that long non-coding RNAs (lncRNAs) are abundant and diverse within fungal genomes, are transcribed from intergenic and genic regions alike, and carry out a number of distinct roles60,61,70. Only recently have fungal lncRNAs been identified and characterized in fungi other than the model yeasts S. cerevisiae and Schizosaccharomyces pombe. These newly characterized fungal lncRNA transcriptomes show little conservation between species as well as little consensus in post-transcriptional modifications71–73. Across fungi, lncRNAs have been shown to regulate gene expression in cis and in trans, influencing mating behaviors, sexual development, morphogenesis, cellular metabolism, virulence, drug resistance, and genomic stability61,70,74. One class of lncRNAs are antisense lncRNAs (ASlncRNAs), which overlap with the open-reading frames of genes in an antisense orientation and regulate expression of their cognate transcripts. It has been shown that loss of RNAi in fungi is correlated with expansion and increased abundance of ASlncRNAs, suggesting that these elements play an important role in modulating gene expression and therefore in the stability and evolution of fungal genomes75–77. Additional deep RNA sequencing among numerous and diverse fungi complemented with molecular genetic analyses will provide further insight into the diversity of lncRNAs and their structure, function, evolution, and impact on overall genomic evolution.
Despite having a conserved role in proper chromosome segregation, centromeres are one of the most rapidly evolving sequences among eukaryotes62. Previous studies focused on centromere identification in relatively few ascomycete fungal species, including S. cerevisiae, S. pombe, and Candida albicans78,79. These studies revealed that centromere length in fungi could range from a few hundred nucleotides to 300 kb in length and varied significantly among related species. These studies also established that centromeres are defined by the binding of a conserved histone H3 variant, CENP-A, along with many other conserved kinetochore proteins. Over the past several years, whole-genome sequencing with long-read Nanopore and PacBio sequencing platforms has led to the identification of centromeres in many other fungi and a better understanding of centromere structure and evolution. One such study revealed that centromeres across the Schizosaccharomyces group harbor a similar architecture but vary in their sequence, suggesting sequence is not critical for centromere function25. Centromeres in the plant-pathogenic ascomycete Magnaporthe oryzae were found to consist of long AT-rich sequences, a feature conserved in related species26.
Characterization of centromeres in the Cryptococcus pathogenic species complex revealed that centromeres in Cryptococcus are long, rich with retrotransposons, and syntenic among different species80,81. However, centromere lengths vary significantly among three pathogenic species in this complex due to the truncation of retroelements as a result of RNAi loss in one of the species81. A more recent study in another basidiomycete group of Malassezia species identified short centromeres (less than 1 kb), which were highly AT-rich and devoid of any repeat content82. Following the pattern in Schizosaccharomyces and the Cryptococcus species complex, centromeres in Malassezia are also syntenic between closely related species.
The nature of centromeres in the Mucoromycota phyla of fungi have also been characterized using Mucor circinelloides, an emerging model organism83. It is important to note that M. circinelloides lacks CENP-A but harbors other conserved kinetochore proteins. Identification of centromeres in this species revealed a mosaic nature of centromeres where kinetochore binding was restricted to a small region, flanked by multiple copies of a retrotransposon. Each of these studies emphasizes the fact that fungal centromeres are highly diverse with few conserved features and are rapidly evolving, even among closely related species. Interestingly, all centromeres identified thus far are in ORF-free regions that are mostly devoid of transcription, and many centromeres are AT-rich in nature. Combining these features together might help in predicting centromeres in fungal species where experimental tools have not been applied or are not yet available.
Other genomic regions in which rapid evolution has been observed are the subtelomeres and the repeat-rich regions of plant pathogens that harbor effector genes mediating virulence. Subtelomeric loci are prone to rapid expansion and contraction and often encode virulence-associated genes as well as biosynthetic gene clusters for many secondary metabolites. The impact of rapid subtelomeric evolution on virulence has been highlighted in the human fungal pathogens Candida auris and Aspergillus fumigatus as well as in the plant fungal pathogen Pyrenophora teres f. teres63–65. Interestingly, in the emerging fungal pathogen C. auris, a particular genetic lineage that has not been involved in outbreaks exhibits high mutation rates and genomic instability, especially in subtelomeric regions. This subtelomeric variation is hypothesized to be attributable to a loss-of-function mutation in DCC1, a gene involved in regulating telomere length, genome instability, and the cohesion of sister chromatids in yeast64,84. Similar to subtelomeric regions, the regions harboring effector genes, also known as effector compartments, in fungal plant pathogens are also prone to frequent expansion and contraction events10,11. These regions are enriched in transposable elements and are regularly located near chromosome rearrangement breakpoints, which contributes to their largely lineage-specific identity11. Effector compartments make up the rapidly evolving portion of the “two-speed” genomes of many plant fungal pathogens. Rapid evolution in these loci is attributed to the expansion, contraction, and recombination mediated by repetitive transposons within the region as well as repeat-induced mutation (RIP) and epigenetic silencing10.
The transmission of alleles between species can have a profound impact on genomic evolution. Instances of inter- and intra-kingdom HGT, genetic introgression following hybridization, and meiotic drive elements have all been shown to be strong forces of genomic evolution and have thus received increased attention over the last decade. Robust whole-genome sequencing of isolates within a species as well as the increasing diversity of fungal species sequenced have allowed for the identification of these allelic transmission events and mechanisms and also shed light on their origins12–14,85–91.
HGT refers to the lateral exchange and integration of genetic material between two species, unlike the vertical inheritance pattern between parent and offspring92. Previously, prokaryotic species were thought to experience frequent HGT events, while relatively few HGT events were thought to have occurred in fungi. However, advances in whole-genome sequencing have, by increasing efficiency and reducing costs, expanded the number of fungal genome sequences available and allowed the identification of numerous HGT events across the fungal kingdom; many instances of HGT events in fungi have been extensively characterized and reviewed16–19. HGT events appear to be more frequent in organisms that have undergone significant niche adaptation such as commensal organisms like the saprophytic skin commensal Malassezia species, which have acquired a bacterial flavohemoglobin as well as many other bacterially derived genes93. Similarly, obligate intracellular pathogens belonging to the Microsporidia and Cryptomycota phyla have experienced significant HGT, in which up to 2% of genes are estimated to be derived from HGT events85. This work has helped to markedly clarify the number of HGT events in Microsporidia because only a small number of HGT events involving bacteria- and animal-derived genes had been previously documented, and these events were thought to be rare16,94–96. In addition to the relatively common HGT of bacterially derived genes in fungi, an exciting new study has identified instances of trans-kingdom genetic transfer between an arbuscular mycorrhizal fungal species and its symbiotic plant partner97. Another case of trans-kingdom HGT was shown in basal fungi (Chytrids) where a key cell-cycle regulator protein evolved from a viral protein86. This study provides key insights into the evolution of cell-cycle regulating machinery that evolved differently in fungi and animals. Further characterization of genomic diversity within and among species across all domains of life will help to identify additional instances of HGT.
Introgression, also known as introgressive hybridization, refers to the integration of genetic alleles from one genetic lineage into another following hybridization. Population-based studies in fungi have revealed the high frequency with which introgression can occur. Two recent examples include 1) the loss and regain of fermentation capacity by Kluyveromyces lactis, which was demonstrated to be a result of introgression of a subtelomeric locus from Kluyveromyces marxianus, and 2) finding that a lichen-forming Rhizoplaca species likely originated from an introgressive hybridization event12,13. In another intriguing study, introgression following hybridization was shown to be the primary force of evolution in the fungal pathogen responsible for Dutch elm disease, driving changes in host–pathogen interactions via introgression of virulence-associated genes and changes in sexual reproduction via introgression of mating-type loci14.
Population genomics approaches have also revealed the prevalence of meiotic drive elements throughout the fungal kingdom. Meiotic drive elements are genetic elements that cause non-Mendelian patterns of inheritance by either biasing their inheritance in sexual progeny or by killing progeny that do not inherit the element, also known as killer meiotic drive elements. Fungal meiotic drive elements were first identified in Neurospora many decades ago98. Subsequent studies have further characterized the meiotic drive elements in Neurospora, known as Spore Killers, and have revealed other Spore Killer meiotic drive elements in Podospora87,88,99–102. Over the past several years, elegant and thorough research in Schizosaccharomyces species has identified several novel meiotic drive elements15,89,90,103,104. Studies have also shown that in addition to the genomic loci that can act as meiotic drive elements, supernumerary chromosomes in the plant pathogen Z. tritici show evidence of meiotic drive91. The ability of genetic elements to bias their inheritance without conferring any adaptive benefits or potentially reducing fitness can greatly influence the genomic landscape and evolutionary trajectory of a species.
Instances of large-scale genomic evolution include chromosomal structural rearrangements and numerical increases or decreases in chromosome copy number. Chromosomal structural changes are rearrangements of chromosome segments that result from recombination or DNA damage repair. Numerical alterations include variation in the chromosome number due to aneuploidy or polyploidy as well as changes due to accessory chromosomes. The past several years have seen a significant increase in studies characterizing such events.
Structural variations or chromosome rearrangements occur when chromosome segments are shuffled across the genome. These structural variations include deletions, inversions, duplications, and translocations. Chromosome rearrangements are known to occur in all eukaryotic genomes105–107. These rearrangements can occur within a single chromosome (intra-chromosomal) as well as across different chromosomes (inter-chromosomal). Many different mechanisms are known to mediate rearrangements, including meiotic recombination, double-stranded break (DSB) repair during mitosis, and transposable elements108,109.
The prevalence and mechanisms of chromosome rearrangements in fungi have been known for a long time, but recent advances in long-read sequencing have highlighted the importance of these events in multiple cellular processes. Several studies comparing the genome evolution of closely related species revealed that chromosome translocations can drive the evolution of secondary metabolite-producing gene clusters, and many of these translocations coincide with transposable elements110–112. Some of these rearrangements are also correlated with gene loss, the generation of genetic diversity, and changes in pathogenicity113. A study in the basal Taphrina genus showed that chromosome rearrangements can drive the evolution of effector superfamily genes and host adaptation as well as speciation114. Another study comparing the genomes of 71 ascomycete species suggests that the rate of chromosome rearrangements accelerated the process of speciation in these species115. A comparison of two different subphyla showed that the number of rearrangements is directly correlated with species richness in each respective subphylum. The same study also showed that whole-genome duplication and pathogenicity might elevate gene order divergence and genome rearrangements115.
Using a direct experimental approach, we recently showed that centromere-mediated chromosome rearrangements, induced by targeting DSBs at centromeric retrotransposons with CRISPR, can alone drive reproductive isolation and thus play a role in speciation116. While centromere-mediated translocation has been proposed to occur based on genome comparisons, we showed its direct impact on the sexual cycle. Such centromere-facilitated chromosome rearrangements have been observed in Malassezia and Candida and are proposed to play a role in both chromosome and karyotypic evolution in these species82,117,118. Centromere-mediated chromosomal translocation has also been proposed as a driving force during the evolution of the mating-type locus in Cryptococcus from an ancestral outcrossing tetrapolar system to a derived bipolar inbreeding state80.
Combined, these studies show that structural variations play a critical role in the evolution of a diverse array of functions in fungal species, including metabolism, pathogenicity, host adaptation, and speciation. Their role in the evolution of secondary metabolite-producing clusters has been observed in multiple fungal species complexes, suggesting that this is a common mechanism via which metabolic potential can be altered or expanded. Additionally, the role of chromosome translocation in speciation has received significant support as a result of these studies. Future studies will reveal the unexplored significance of these chromosome structural changes in fungi. One key aspect to focus on will be the impact of these changes on pathogenicity and how they contribute towards the evolution of virulence in fungal pathogens.
Whole-genome duplication or polyploidy refers to scenarios in which a cell or organism possesses three or more complete sets of chromosomes compared to the standard haploid karyotype. Polyploidy is found across the fungal kingdom, and one route to its generation involves whole-genome duplication23,24. Indeed, analysis of the whole-genome sequence of Saccharomyces revealed that one such whole-genome duplication event was responsible for the evolution of this genus and likely involved an interspecies hybridization event30,119. Despite the presence of polyploidy across the fungal kingdom, studies analyzing the polyploidy of fungal genomes have started to re-emerge23. Many clinically relevant fungi show variation in ploidy, mainly in response to stressful or new environmental conditions. Some fungi can also exist in multiple different ploidy states and exhibit ploidy transitions from one state to another. Studies in S. cerevisiae, as well as C. albicans, found that ploidy variation can drive rapid adaptation in response to environmental conditions and vice versa120,121.
A well-studied process of ploidy transition is the parasexual cycle in C. albicans122. During this process, tetraploid C. albicans cells undergo ploidy reduction to generate more stable diploid cells, which can be selectively advantageous123. This process is accompanied by concerted chromosome loss and generates aneuploidy124. Recent studies have provided intriguing insights into this process. Tetraploid C. albicans cells were found to be metabolically hyperactive, generating reactive oxygen species and DNA DSBs, resulting in increased genomic instability125. Another study found that the genome reduction in this parasexual cycle involves recombination, similar to meiosis observed in other organisms, and thus has been termed para-meiosis126. A closely related species, Candida tropicalis, was found to exhibit a combination of same- and opposite-sex mating to generate polyploidy, which in turn increases genetic diversity127.
Another human fungal pathogen, C. neoformans, has been shown to exist in haploid or hybrid diploid states. C. neoformans can also exist in a polyploid state, known as titan cells, produced in response to host conditions128,129. These titan cells produce haploid and aneuploid progeny and thereby enhance stress adaptation130. A concerted effort from three independent studies established that this phenotype can be induced by multiple environmental as well as genetic signals131–133. Similar to C. albicans, the ploidy reduction in these titan cells is mediated by the activation of meiotic genes134. While these studies explore the reduction of a polyploid genome to a more stable state, the exact role of polyploidy and the factors responsible for inducing polyploidy are not well understood. The presence of different levels of ploidy in S. cerevisiae natural isolates hints toward its role in adaptation to environmental conditions, but such natural resources have not been explored in other fungal species yet.
Aneuploidy refers to the stage when a cell deviates from its balanced genome by gaining or losing chromosomes. Aneuploidy has been described in a large number of fungi but mainly with respect to stress conditions. While aneuploidy is commonly thought of as a deleterious state, and in humans is invariably associated with disease states or is lethal, many studies have found naturally occurring aneuploid isolates in different fungal species20,24. It has been proposed that aneuploidy may represent an intermediate, transient state that can promote evolutionary adaptation135,136. Experimental evolution of multiple S. cerevisiae strains showed that aneuploidy frequently occurs during the course of evolution137. Similar results were observed when 47 non-laboratory isolates of S. cerevisiae were analyzed138. The gene SSD1 and its contributions to mitochondrial physiology were identified as the genetic basis of aneuploidy tolerance in these natural isolates139. A genome-wide genetic deletion screen in S. cerevisiae found that genes involved in a ubiquitin-mediated proteasomal degradation pathway also confer tolerance to aneuploidy140,141.
Aneuploidy has been previously known to play a crucial role in drug resistance in pathogenic fungi like C. albicans and C. neoformans142–144. In both cases, drug resistance involved amplification of fluconazole target genes like ERG11 and genes responsible for the expression of drug efflux pumps, either by isochromosome formation in C. albicans or aneuploidy in C. neoformans. Newer studies have shown that aneuploidy can play roles in osmotic stress, flocculation, and ethanol survival as well as responses to starvation conditions in S. cerevisiae145–147. We have also shown that aneuploidy can be essential to overcome reproductive barriers between two strains harboring chromosomal translocations116. Overall, aneuploidy seems to be a conserved fungal response to stress or harsh environmental conditions. However, the advances discussed here reveal a role for aneuploidy in adaptation to growth conditions as well as pathogenicity in fungal species. With their well-characterized role in drug resistance, mechanisms leading to aneuploidy also present potent targets for drug development.
Accessory, supernumerary, or B chromosomes are all different names for extra chromosomes that are present in a cell beyond the reference karyotype. These extra chromosomes segregate in a non-Mendelian fashion, are dispensable, and do not undergo recombination or pairing with the main, essential (also known as A) chromosomes21,22. Accessory chromosomes are well studied in animals and plants but have also been observed in several plant fungal pathogens. Earlier reports suggested that these chromosomes are required for virulence and can be horizontally transmitted148,149. More recent studies have focused on the characterization of these chromosomes at the molecular level in Z. tritici and M. oryzae. In Z. tritici, B chromosomes were found to be epigenetically indistinguishable from the core set of chromosomes, whereas the B chromosome (also known as a mini-chromosome) in M. oryzae was found to be more enriched with transposons relative to the core chromosomes150,151. B chromosomes in both of these species were also found to harbor active centromeres26,150. However, the B chromosomes in Z. tritici are unstable and frequently lost during vegetative growth152.
Interestingly, the stability of B chromosomes also depends on the host genotype and thus plays a vital role in host–pathogen interactions153. The mini-chromosome in M. oryzae was proposed to be involved in the shuffling of AVR effector genes between the mini-chromosome and core chromosomes, thus aiding in host adaptation151. While the exact function of genes present on these accessory chromosomes is still unknown, they significantly influence the pathogenicity of the fungi in which they are present. For this purpose, an in-depth characterization of accessory chromosomes is essential and should include genetic as well as epigenetic approaches. The identification of B chromosomes in more species will advance the field and may reveal new insights into their roles in pathogenesis, adaptation, evolution, and possibly speciation.
A multitude of advances in high-throughput sequencing, also known as next-generation sequencing, and molecular biology methods have helped to reveal and characterize the forces driving evolution in fungal genomes. Efficiency and improved affordability of high-throughput whole-genome sequencing on Illumina sequencing platforms have promoted some of the largest advances in our understanding of fungal genome evolution by enabling the sequencing of numerous strains within a single species. The whole-genome data of various isolates within a species provides a pan-genome resource that spans the population variation within a species and also provides an opportunity to assess genomic microevolution and potentially macroevolution from a population genetics and genomics approach. Pan-genome sequencing has been especially helpful for identifying rates and dynamics of intraspecific genomic exchange, including the evolution and propagation of meiotic drive elements, as well as identifying inheritance of interspecific genetic material, including HGT events and instances of hybridization leading to introgression.
The technological advancements of the long-read PacBio and Oxford Nanopore Technologies sequencing platforms have been critical in both further understanding macroevolution in fungi and significantly advancing the identification and characterization of chromosome structural variations. Using these technologies, many fungal genomes were sequenced and compared to develop synteny plots, and these synteny plots have allowed the detection of chromosome translocations. Some of these translocations were found to result in loss or gain of effector genes and also to have contributed to the evolution of secondary metabolite genes. A striking example of the advantages long-read sequencing technologies can have over short-read sequencing techniques was recently demonstrated in S. pombe, in which PacBio and Nanopore sequencing platforms were able to accurately predict 16-fold more genomic structural variations than short-read technologies27,28. Additionally, long sequencing reads often span repetitive regions and transposons, allowing sufficient coverage of unique flanking regions, which is essential for assembling repeat-rich regions of genomes. This particular feature has allowed the identification of centromeres in many fungal species, especially in cases where centromeres harbor repetitive elements or transposons. Another significant feature of long-read sequencing methods is the ability to capture telomere repeat sequences, thus allowing telomere-to-telomere genome assembly. The identification of telomere repeat sequences at the ends of mini-chromosomes or B chromosomes provided support for their existence and helped differentiate these supernumerary elements from core chromosomes. Similarly, it was also helpful in the determination of chromosome number in several species. Another type of technique that has helped to achieve enhanced fungal genome assemblies is chromosome conformation capture, which includes methods such as Hi-C154,155. By defining the interactions between DNA sequences, Hi-C has aided in resolving genome assembly conflicts, thus improving assemblies156,157. It is important to note that these long-read sequencing techniques are highly error-prone and thus need to be corrected with less error-prone short-read Illumina sequencing prior to gene prediction or gene calling for annotation.
Research over the last decade has provided unprecedented insight into the evolution of fungal genomes from both micro- and macro-evolutionary perspectives. The ease, efficiency, and affordability of sequencing combined with great community efforts have promoted research and given rise to findings that illuminate how small-scale genomic changes such as hypermutation, mobilization of various genetic elements, allelic transmission, and rapidly evolving regions can impact the evolution of fungi. Due to advances in long-range whole-genome sequencing technologies and the generation of telomere-to-telomere assemblies, researchers can now characterize the evolution of ploidy and chromosome structure, particularly in highly repetitive regions, with high accuracy and confidence. Advances in understanding fungal genome evolution at the nucleotide, gene, chromosomal, and nuclear levels are illustrated in Figure 1. Long-read sequencing platforms will continue to be important technologies for further characterization of highly repetitive regions, such as centromeres, transposon-rich regions, subtelomeric loci, and telomeric repeats. Studies employing these techniques in a vast array of species will shed light on evolutionarily missing links while also providing a better understanding of already existing processes.
Methods and techniques that attempt to further improve long-range sequencing and its accuracy as well as resolve artifacts and biases generated through library preparation, sequencing itself, and subsequent analyses will also improve our understanding of fungal genome evolution.
Improvements are still especially needed in RNA sequencing and sequencing library preparation. Because of the high abundance of some RNA species, like ribosomal and translational RNAs, it remains difficult to detect rare RNA species, such as non-coding RNAs and alternatively spliced transcripts. Developments on this front will aid in efforts to further identify and characterize lncRNA transcriptomes in fungi and can help to determine how lncRNAs vary at the pan-genome level and influence genomic evolution.
Continued progress with sequencing and other genomic technologies will provide more insight into the driving forces and consequences of genomic evolution. Additional sampling and sequencing of novel fungi and less well-characterized species will provide further insight into genomic diversity in the fungal kingdom. Owing to the variety of fungal niches and lifestyles, sufficiently culturing fungi for sequencing efforts can be tedious and often impossible based on current knowledge and techniques. Therefore, advances in single-cell sequencing methods could be highly advantageous to characterize the genomes of unculturable species or species that are difficult to isolate. This will also help the sequencing and study of fungi beyond Ascomycota and Basidiomycota, which have been the focus of the majority of research until now. Studies involving fungi from Zygomycota and Chytridiomycota are only beginning and are already providing exciting insights into the diversity and uniqueness of these fungal phyla.
In addition to the analyses conducted on the nuclear genomes presented in many of the studies discussed here, it will be interesting to conduct population genomics studies on mitochondrial genomes in parallel to characterize the diversity, inheritance dynamics, and evolution of these important organelles. Research focused on identifying and characterizing fungal-associated viruses, or mycoviruses, is also garnering increased attention, and developments on this subject will be interesting, especially with regard to their propagation dynamics and impacts on host genome evolution. These potential future research avenues present great and exciting opportunities to further advance the field of fungal genetics.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)