Keywords
Vaccine Design, Immunoinformatics, Rift Valley fever virus, Immune Simulation
Vaccine Design, Immunoinformatics, Rift Valley fever virus, Immune Simulation
2D, two-dimensional; 3D, three-dimensional; ACC, auto cross co-variance; CAI, codon adaptation index; CTL, cytotoxic T-lymphocyte; E. coli, Escherichia coli; GC, guanine and cytosine; GRAVY, grand average of hydropathicity; HTL, helper T-lymphocyte; IEDB, immune epitope database; IFN, interferon; IFN-γ, interferon-gamma; IL, interleukin; IL-10, interleukin 10; IL-4, interleukin 4; kNN, k-nearest neighbors; LBL, linear B-lymphocyte; MHC, major histocompatibility complex; NCBI, National Center for Biotechnology Information; NMA, normal mode analysis; PDB, Protein Data Bank; PI, isoelectric point; RMSD, root mean square deviation; RVF, Rift Valley fever; RVFV, Rift Valley fever virus; SVM, support vector machine; TAP, transporter associated with antigen processing; TLR4, toll-like receptor 4.
Rift Valley fever virus (RVFV) is a risk to worldwide public health and farming, especially in parts of Africa, Madagascar, and the Middle East1. RVFV epidemics have killed hundreds of thousands of animals, more than a thousand humans, and caused significant economic losses2. RVFV is a negative sense, single-stranded RNA (ssRNA) virus3 and belongs to the family of Bunyaviridae. RVFV was first identified in Kenya among sheep, in the vicinity of Lake Naivasha4. Using mosquitoes as a vector it can cause large scale transmission, causing mild symptoms like fever, back pain and nausea to fatal illnesses including critical eye diseases, encephalitis in humans, and lethal hemorrhage in animals5. Mortality of up to 90% has been reported in newborn animals and up to 30% in adult animals6, although the mortality rate for humans has been reported to be approximately 2%7. Epidemic alarms have persuaded several national and international health organizations to issue cautions about the rising risk of infection in Rift Valley fever (RVF) uninfected countries, like Europe and USA, due to the existence of vectors of transmission which are highly permissive, further compounded by global animal trade7–9. These reports unanimously concluded that coordinated efforts are needed in order to prepare for preventive measures against the recurrent emergence of RVFV.
RVF is an arthropod-borne zoonotic infectious viral disease caused by RVFV9. Direct contact via tissue, body fluids or blood of RVFV infected animals acts as the prime mode of transmission for humans10. Mosquitoes are a major vector for RVFV spread11, infecting humans over long distances and even causing vertical transmission between livestock8. The incubation period for RVFV is about 2–6 days in humans. The virus consists of a negative sense, triple segmented (large, medium, and small) ssRNA molecule and has a viral genome encoding four proteins: glycoprotein, RNA-dependent RNA polymerase, non-structural protein, and nucleocapsid protein12–14. Although the non-structural proteins facilitate RVF to survive inside its host by inhibiting first-line immunogenic responses, its glycoproteins are essential and highly crucial for invasion, entry and viral replication inside the host cell13,14. Thus, the viral glycoproteins were targeted for our multiepitope vaccine design, which would be constructed using glycoprotein epitope sequences evoking an immune response inside the human system13,14. At present, epitope-based candidate vaccine design against viruses and bacteria as well as parasites has become very popular and has been done previously15–20. Multiepitope vaccines consist of short peptide fragments of immunogenic stimulants, which trigger a strong immune response and allow for a significantly lower chance of allergenic reactions inside the host system21. The identification of immunogenic epitopes derived from viral glycoprotein or nucleocapsid sequences has significantly enhanced the in silico development of peptide vaccines22.
In our study, we screened the RVFV proteome to find the highest antigenic glycoprotein to predict T and B cell epitopes using a computational approach. Subsequently, cytotoxic T-lymphocyte (CTL), helper T-lymphocyte (HTL) and linear B-lymphocyte (LBL) epitopes predicted to be immunogenic and antigenic were shortlisted, which were further subjected to toxicity and allergenicity analysis. A vaccine design was assembled by combining all the assessed CTL, HTL and LBL epitopes using linkers and an appropriate adjuvant. Physicochemical analysis and solubility prediction were performed in Escherichia coli (E. coli) to assess the vaccine. Next, secondary and tertiary vaccine models were predicted using structure analysis tools. The predicted tertiary structure was refined and validated. Moreover, a disulfide bond was introduced by disulfide engineering, improving vaccine stability. Interactions within the vaccine-TLR4 complex were studied using molecular docking and evaluated using molecular dynamics simulation. Additionally, codon optimization was carried out to increase the translation efficiency of the designed vaccine within a E. coli K12 host. Finally, immune simulation was carried out to predict real-life immunogenic potency of the vaccine. The employed steps for the development of vaccine are summarized in Figure 1.

LBL, linear B-lymphocyte; HTL, helper T-lymphocyte; CTL, cytotoxic T-lymphocyte.
The 232 complete proteomes of RVFV were obtained from ViPR (Virus Pathogen Resource) database (Supplementary Table 1, Extended data)23. ViPR is a reliable and open database for pathogenic virus families24. The viral proteome sequences retrieved were evaluated using VaxiJen v2.0 server for scores indicating antigenic influence. The threshold for antigenicity was fixed at 0.525. VaxiJen v2.0 sever possesses high prediction powers and utilizes auto cross-covariance (ACC) transformation methods. The glycoprotein sequence with the highest antigenicity score was taken from this proteome for further analysis.
the NetCTL v1.2 tool was utilized to isolate CTL peptides from the RVF glycoprotein sequence. This server generates different nonamer epitopes against 12 supertypes (A1, A2, A3, A24, A26, B7, B8, B27, B39, B44, B58, B62). NetCTL v1.2 identifies CTL epitopes depending on C-terminal cleavage, TAP competence and MHC-1 complex binding26. The threshold was determined at 0.5 with a corresponding sensitivity of 0.89 and specificity of 0.94. The MHC-I affinity for the CTL peptides was identified using the MHC-I IEDB web server27 and a consensus percentile of ≤5 was set to narrow down CTL epitopes, for obtaining high-affinity epitopes with MHC-1 alleles.
The isolated CTL epitopes were assessed for antigenicity by submitting them to the VaxiJen v2.0 server. To ensure proper induction of immune response in the human body, the immunogenicity of these epitopes was evaluated using the IEDB Immunogenicity tool28. Furthermore, AllerTOP v2.0 server was chosen for predicting allergenicity and ensure that the vaccine construct does not induce an allergic reaction in humans. AllerTOP v.2.0 uses k-nearest neighbors (kNN) methods to distinguish allergens from non-allergens29. Lastly, the selected nonamers were screened for probable toxicity using ToxinPred server, which predicts toxic epitopes using quantitative matrix methods and machine learning technology30.
HTL acts as the orchestrator to stimulate B cells, macrophages, and CD8+ cells to fight against pathogens. Consequently, HTL epitopes are important for making an effective vaccine31. HTL epitopes, each 15-mer in length, were identified by utilizing the IEDB MHC-II tool. The anticipation of binding of the epitopes to class II alleles, HLA-DP, HLA-DR and HLA-DQ, were determined utilizing the consensus 2.22 prediction method on the same server32. Due to obtaining a large number of epitopes, a percentile rank of ≤0.3 was set as a threshold.
HTLs release cytokines such as interferon-gamma (IFN-γ), interleukin 4 (IL-4), and interleukin 10 (IL-10) that can activate immune cells in the body. In addition, cytokines released by HTLs can survive past inflammatory responses and avert tissue damage. Therefore, HTL epitopes that can induce the release of cytokines are important in vaccine development. Therefore, in order to incorporate the epitopes that induce IFN- γ, we used the IFNepitope server, through the hybrid method (motif and SVM)33. Furthermore, the peptides were evaluated to check if they induce IL-4 and IL-10 using IL4pred and IL10pred servers, respectively34,35.
The activation of B cells plays a vital part in the activation of the humoral immune response and generation of plasma cells against a specific antigen, and distinguishing LBL epitopes is another vital step in the development of epitope-based vaccine constructs. LBL epitopes were predicted using a combinatorial algorithm of gradient boosting and a randomized tree method using the iBCE-EL server36. The predicted LBL epitopes were then reevaluated for their antigenicity score with VaxiJen v2.0 server, and non-allergenic and non-toxic epitopes were predicted using the AllerTOP v2.0 server and ToxinPred tool, respectively.
For an epitope to be antigenic and evoke a strong immune reaction it needs to be acknowledged by the MHC complex molecule. MHC alleles are the most polymorphic and occur in thousands of HLA combinations in humans. Therefore, an HLA allele with a high frequency of occurrences in the majority population of the world would have a high chance of exerting an effect immunogenic37. In our study, we wanted to find out the distribution, presence and frequency of the T cell epitopes, which were selected for the purpose of designing the vaccine structure. Allele Population Coverage of IEDB population coverage tool was utilized for the calculation of population coverage38. A prediction analysis was run on regions of Africa where the RVFV outbreak initially started and the neighboring countries that were mostly affected and also across the entire world.
All the predicted and assessed epitopes i.e. CTL, LBL, and HTL peptides were joined together using linkers and an adjuvant sequence was added upstream of all of them to form a vaccine structure. 50S ribosomal protein L7/L12 (GenPept Accession: P9WHE3) is a toll-like receptor 4 (TLR4) agonist and was chosen as an adjuvant for the construct to boost the immune response against it21. TLR4 can recognize viral glycoproteins and bacterial ligands39,40 stimulates the production of cytokines against them41. The CTL peptides and the 50S L7/L12 protein adjuvant were linked together with the EAAAK linker to ensure adequate separation between each component for their effective interaction with their respective targets. This linker was chosen as it separates bifunctional fused protein domains42. Each CTL epitope was separated using AAY linkers, and HTL epitopes by GPGPG linkers. Independent immunogenic potential of each LBL epitope was preserved by separating them using KK linkers43.
To avoid the risk of elicitation of autoimmunity by molecular mimicry, we screened the vaccine construct against the Homo sapiens protein sequences (NCBI: txid9606) through the use of NCBI BLASTp44.
Vaccines are developed to provide protection against diseases by evoking an immune response after injection against specific antigens, without causing any disease. Thus, vaccines need to have antigen inducing capability without being allergenic to the receiver. Therefore, the multiepitope vaccine construct was tested for antigenicity and allergenicity in each step of the initial design, using VaxiJen v2.025 and AllerTOP v2.0 servers, respectively29. Predictive assessment of different physiochemical attributes of the subunit vaccine was carried out using the ProtParam server45. Lastly, the SOLpro server in the SCRATCH suite was utilized to assess the solubility of the vaccine structure in E. coli, with a view to determining the bioavailability of the vaccine46.
Predicted two dimensional (2D) configuration of our vaccine design was generated using the PSIPRED v4.0 web tool. The PSIPRED tool utilizes the query amino acid residues to predict a 2D model using two-feed-forward neural networking along with PSI-BLAST47. The three dimensional (3D) model of our designed vaccine was attained through the I-TASSER web tool, which exhibits protein modeling via a hierarchical procedure to estimate and bring about suitable structure and function48. The I-TASSER site helps to generate 3D structure of a protein and determine its functions using a state-of-the-art algorithm with high precision. This web tool enables determination of C-score, TM-score value and root mean square deviation (RMSD), along with the top five predicted structure models of the given protein sequence48. The produced 3D structure was chosen based on its C-score value and downloaded in PDB format. The server provides a C-score ranging between -5 to 2, where a higher value indicates a better model48.
Our 3D vaccine structure was refined using the GalaxyRefine server. The web-based program reconstructs sidechains, then repacks them using dynamic simulations for proper structural relaxation49. The ProSA tool was used to recognize possible errors in the predicted tertiary model and for structural validation16. If the calculated score of ProSA deviates from the given range, this indicates that the primary sequence and predicted structure possibly contain flaws50. Additionally, the Verify3D server was employed to examine the congruity of a tertiary structure with its primary sequence. It is done by specifying a basic grade depending upon its position and surroundings and correlating the findings to known verified structures51. Ramachandran plot generation was carried out using the RAMPAGE tool52. The Ramachandran analysis plot is a visual representation of energetically permitted and rejected dihedral planar angles based on weak Van der Walls force of interactions between amino acids of the side chain. The RAMPAGE score enumerates the residues residing in favored, allowed and disallowed regions (in %) based on the PROCHECK principle53.
Disulfide bonds are intra-protein bonds that can help stabilize tertiary or quaternary interactions in a protein54. Therefore, disulfide engineering was accomplished by introducing two novel bonds in the vaccine framework by employing the Disulfide by Design v2.12 web tool. The refined vaccine complex was uploaded, checked for a residue-pair match within parameters of χ3 angle -87° or +97° ± 30 and Cα-Cβ-Sγ angle 114.6° ± 10. Appropriate amino acid duos were chosen and mutated to cysteine residues55.
Codon refinement is a vital step for the expression of the protein inside the host system. Therefore, codon adaptation was carried out by utilizing the JCat web tool in order to improve protein translation. E.coli K12 was selected for cloning since unadjusted codon sequences will reduce protein expression56. Termination of transcription (rho-independent), prokaryotic ribosome region of attachment and restriction enzyme cut-off sites were disregarded by setting the parameters to “avoid”. The server further provided two additional measures such as the codon adaptation index (CAI) and GC content of the refined sequences57. The codon nucleotide adapted sequence was cloned using SnapGene v5.0.8 software into a E. coli pET28a(+) vector obtained from SnapGene58. ApE and Genome Compiler are open access alternative to this software that could be used for this purpose.
Molecular docking is a computerized process that evaluates and assesses the contact between a protein and its receptor. The binding affinity and interaction between the proteins provide a simulated score59. TLR4 can recognize viral glycoprotein60 and therefore, the TLR4 structure was obtained from PDB (PDB ID: 4G8A) and was chosen as a receptor to dock with the refined vaccine61. ClusPro v2.0 web docking server was appointed to determine the binding position and inclination for binding between the designed vaccine and TLR4 receptor62. The vaccine-receptor composite was selected based on the binding position (on active site), docking efficiency and the lowest energy scoring of the docked complexes.
Determining the stability and firmness of the protein-receptor docked structure by molecular dynamics is essential for carrying out an in silico study. Protein stability was resolved by contrasting protein dynamics and their counterpart normal modes63,64. the iMODS tool was used to evaluate and plot amino acid residues and their motion within their inner order via normal mode analysis (NMA)65. The NMA tool surfaced the extent of motion of the vaccine-receptor complex in terms of covariance and ability to deform; along with eigenvalues and B-factor results. The deformability of the complex depends on whether it can rotate each of its residues while staying in its position. The eigenvalue demonstrates the rigidity of motion, being directly proportional to the energy required for the deformation of the structure. Lower eigenvalues represent easier structural deformation65,66.
Simulations of immune response of the vaccine design were executed with the C-IMMSIM v10.1 server. This server uses position-specific scoring matrix (PSSM) and AI-based technology to predict the intensity of immune reaction caused by the multiepitope vaccine67. It evaluates the immunologic response of the subunit vaccine according to an in vivo system and can also simulate immune response through its agent-based dynamic system. The minimum recommended interval is at least four weeks between the first and second dose68. Time-steps for each vaccine dose were fixed: 1, 84 and finally 168 respectively, with an interval of eight hours in-between. Analysis of three doses was conducted in respect to immune cell rise and activity with at least four weeks in between each of the dose sessions.
Amino acid sequences of a total of 232 RVFV proteins were collected from the ViPR database. The VaxiJen v2.0 server revealed the two proteins with the highest antigenicity (Table 1). The glycoprotein sequence was selected for further analysis.
NetCTL v1.2 server predicted 135 unique 9-mer CTL epitopes in total. Out of them, 27 epitopes were predicted as having the ability to act as both antigen and immunogen and were also nontoxic and nonallergenic (Supplementary Table 2, Extended data)23. Only six CTL peptides were shortlisted for the ultimate vaccine design based on a combined score (Table 2).
Similarly, 182 15-mer HTL epitopes and their MHC-II binding alleles were predicted and evaluated by utilizing the IEDB MHC-II tool. Next, cytokine production capability of the selected HTL epitopes was predicted for IL-10, IL-4 and IFN-γ. Only 14 epitopes were found to be capable of inducing any of the three cytokines’ production and to have antigenicity (Supplementary Table 3, Extended data)23. Of those that were capable of producing IFN-γ, two epitopes were chosen with the ability to produce IL-4 but not IL-10 and one epitope was chosen with the ability to produce IL-10 but not IL-4. Therefore, three epitopes were finally shortlisted prioritizing for IFN-γ and IL-4 production (Table 3).
B cells act as antigen-presenting cells that recognize epitopes present on a protein and can bring about a humoral immune response69. The iBCE-EL sever program was used to predict unique LBL epitopes from the RVFV glycoprotein sequence. A total of 310 LBL epitopes were found. After further evaluation, 35 unique epitopes were found to be non-allergenic and non-toxic (Supplementary Table 4, Extended data)23. Amidst those, only five LBL epitopes were chosen for vaccine construction based on iBCE-EL server’s probability scores. LBL epitopes with higher probability scores were chosen. (Table 4).
| CTL epitope | C-score * | Antigenicity | Immunogenicity | Allergenicity | Toxicity |
|---|---|---|---|---|---|
| VFALAPVVF | 1.7143 | Yes | Yes | No | Non-Toxic |
| STAHEVVPF | 1.7042 | Yes | Yes | No | Non-Toxic |
| KLTLEITDF | 1.627 | Yes | Yes | No | Non-Toxic |
| RDNETSAEF | 1.553 | Yes | Yes | No | Non-Toxic |
| FSSVAIICL | 1.3561 | Yes | Yes | No | Non-Toxic |
| ALSIGLFFL | 1.3051 | Yes | Yes | No | Non-Toxic |
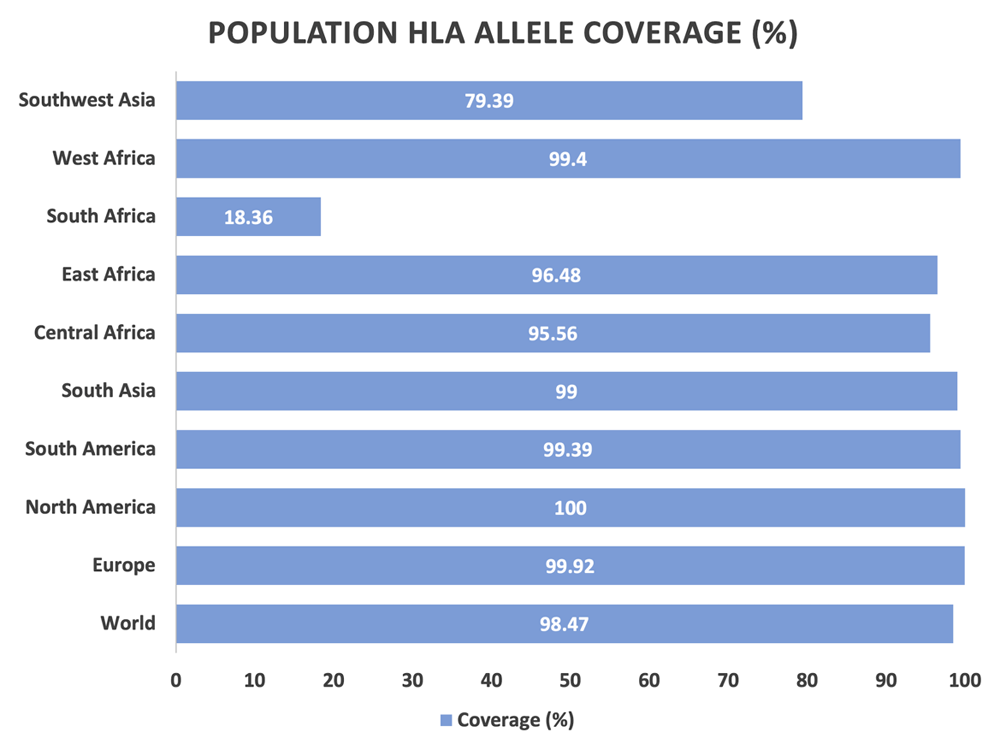
The CTL and HTL epitopes across African regions and Southwest Asia have a mean population coverage of 77.39 %, with an average hit of 1.78. The total combined class 1 and 2 epitopes have significant coverage across Europe (99.92%), North America (100%), South America (99.39%) and South Asia (99%), which indicates the multiepitope vaccine would be a good candidate in eliciting an immune response in these worldwide regions. In the African population, where the initial outbreak began, there is good coverage in the majority of regions, except for South Africa (18.36%), as shown in Figure 2.
To design our multiepitope vaccine, we picked four CTL epitopes (Table 2) based on their high immunogenicity and antigenicity scores and which were non-allergic and nontoxic. For the HTL epitope selection, we looked for epitopes that could produce all three types of cytokine; however, no epitope was found that had the capacity to induce all three types of cytokines (IFN-γ, IL-10, and IL-4). Hence, only two HTL epitopes were chosen from the glycoprotein sequence, which were capable of inducing IFN-γ and IL-4 (Table 3). Three LBL epitopes that were nontoxic and non-allergic and had the best iBCE-EL predicted probability, high antigenicity scores were selected (Table 4). Two extra epitopes were shortlisted for each of the three cases for the purpose of randomization to find the optimum vaccine construct. The choice of adjuvant was 50S ribosomal protein L7/L12, retrieved from the NCBI Protein database (Accession no. P9WHE3) and was linked to the selected epitopes using an EAAAK linker. For the final vaccine construct, four out of six CTL, two out of four HTL and three out of five LBL shortlisted epitopes were chosen based on different combinations and randomization to generate seven potential vaccine candidates, provided in Supplementary Table 5 (see Extended data)23, and were merged with AAY, GPGPG and KK linkers, respectively (Figure 3). The prospective vaccine with the ideal physicochemical property was chosen. The final assembled vaccine is 262 amino acid residues long.
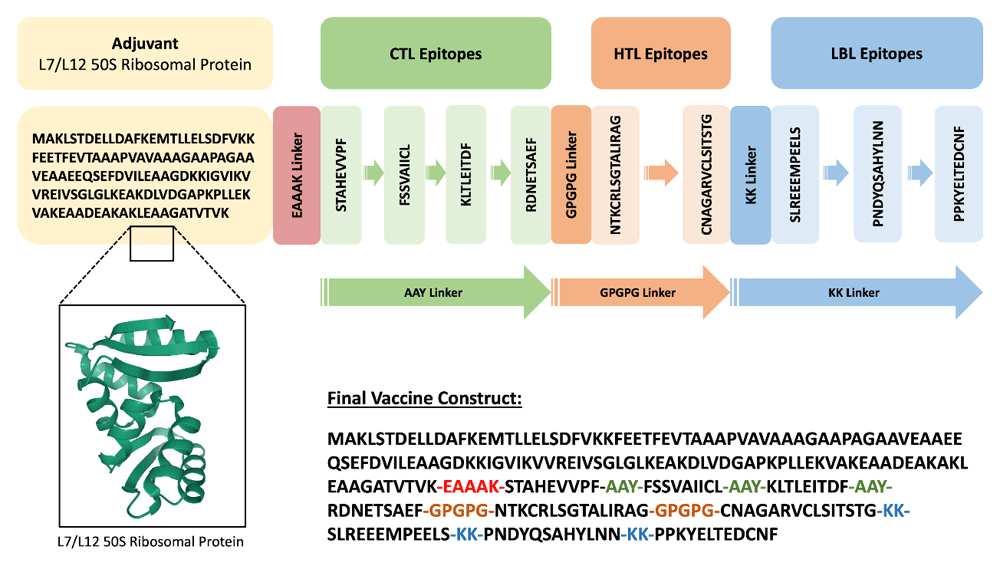
The EAAAK linker was utilized to join the adjuvant at the 5’ end to the rest of the vaccine. CTL epitopes linked with AAY linkers, HTL epitopes linked with GPGPG linkers and form a bridge with the last CTL epitope and first HTL epitope. KK linkers were used to connect LBL epitopes and also form a bridge between the last HTL epitope and the first LBL epitope.
LBL, linear B-lymphocyte; HTL, helper T-lymphocyte; CTL, cytotoxic T-lymphocyte.
Evaluation of the multiepitope vaccine protein for similarity against the Homo sapiens proteome was done using BLASTp to ensure safety. No similar proteins were found, which negates the chance of autoimmunity as a result of the vaccine.
Physiochemical attributes of the vaccine structure were assessed using the ProtParam tool. The vaccine’s molecular weight was estimated to be ~28 kD and showed good antigenic characteristics. The predicted Isoelectric point (pI) of the designed vaccine is 4.96, indicating an acidic nature, and it has an instability index of 27.79, suggesting that the vaccine will maintain good stability inside the host system. The aliphatic index is 84.73, denoting the thermostable nature of the vaccine. The hydrophilic nature of the vaccine was estimated by the grand average of hydropathicity (GRAVY) score of -0.125. Half-life (t1/2) was calculated to be greater than 30 hours in a mammalian reticulocyte, greater than 20 hours in yeast, and more than 10 hours in E. coli. The overall vaccine construct is not allergenic and demonstrated good solubility as shown by the SOLpro server. The above-mentioned assessments suggest that our multiepitope vaccine design has the potential to be a good candidate for RVFV (Table 5).
The 2D structure of the vaccine design was acquired through the PSIPRED v4.0 workbench. The secondary structure consisted of 49.62% α-helices (130 amino acids), 11.83% β-strands (31 amino acids) and 38.55% random coils (101 amino acids) (Figure 4).

Herein, α-helix, β-strands, and random coils are represented with pink, yellow and blue colors, respectively.
The multiepitope vaccine was speculated and constructed using I-TASSER server. The PDB structure (PDB ID: 1DD4) was regarded as the best template for modeling. This server carries out 3D modeling based on the consequence of threading template alignment and simulations are run by merging parameters of the structure assembly. It ranks the confidence of models quantitatively on C-score. The obtained C-score for the modeled 3D vaccine structure was -4.16. The refinement of the 3D structure was carried out by the GalaxyRefine web-tool. The refined and polished model was used to generate a Ramachandran plot, which showed an increased percentage of residues in the favored region. The refined vaccine construct showed 90.0% residues in the favorable region in Ramachandran plot. The global distance test (GDT-HA) score was found to be 0.9055, RMSD was 0.534, MolProbity was 2.209, clash score was 14.2 and poor rotamers were non-existent (Figure 5).

The refined tertiary structure was validated using the RAMPAGE server, ProSA-Web tool and Verify3D server. Validation carried out by the RAMPAGE server using a Ramachandran plot of the unrefined 3D structure showed 69.2% of residues in the favorable region, 20.8% in acceptable regions, and 10.0% of the residues in disallowed regions. After structural refinement was carried out, RAMPAGE revealed 90.4% of the vaccine’s residues resided in favorable regions, and 6.2% and 3.5% of residues were found in acceptable and disallowed regions, respectively (Figure 6A). The ProSA-web and Verify3D (Figure 7) servers were used to validate the unrefined tertiary structure. After refinement, ProSA-web revealed a Z-score of -6.2 for the best vaccine protein model, implying equivalence to the native protein conformation (Figure 6B).

(A) Ramachandran plot analysis of the refined model showing favored, allowed and disallowed regions are 90.4%, 6.2% and 3.5%, respectively. (B) Validation using ProSA web tool, revealing a Z-score of -6.2.

Disulfide bridging was used to stabilize the refined vaccine model. The introduction of cysteine residues was carried out via Disulfide by Design v2.12. Although 23 potential pairs of residues were found (Supplementary Table 7, Extended data)23 suitable for disulfide engineering, only two pairs of residues were chosen based on the energy of binding and χ3 angle. Accordingly, two mutations were created on a pair of selected amino acids, based on the lowest kcal/mol value. For, Leu67-Ala100 residues, the energy value was 2.40 kcal/mol and the χ3 angle was -103.13 degrees and for Phe28-Ala37 duos, the χ3 angle was -111.70 degrees and energy value was 2.86 kcal/mol (Figure 8).
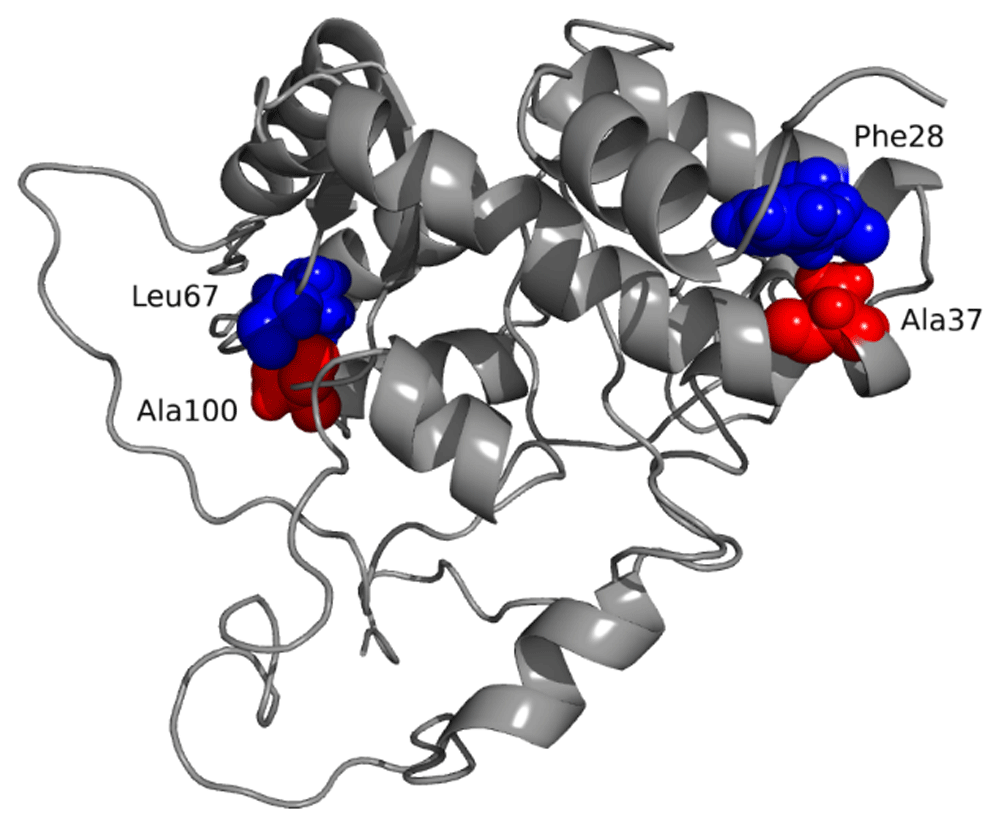
The engineered pairs are indicated by blue and red colors.
The primary objective is to express our constructed multiepitope vaccine sequence using in silico cloning into the E. coli expression system. As such, the subunit vaccine construct requires codon optimization according to the codon usage for the expression pattern system in E. coli. To optimize for maximal expression of our vaccine design in E. coli K12, the JCat tool was used. The optimized codon sequence was 786 base pairs in length. The GC content was 49.75%, which lies within the ideal range of 30–70%. Codon Adaptation Index was 1.0, which is also within the ideal range (0.8–1.0). This ensures the efficient expression of the multiepitope vaccine inside E. coli. XhoI and BamHI restriction sites were inserted into E. coli plasmid pET28a(+) and the adapted vaccine’s codon sequences were inserted into a recombinant E. coli pET28a(+) plasmid using SnapGene software v5.0.8 (Figure 9). The final length of the recombinant construct is 6121 bp.
Outer dark margin represents the vector skeleton.
The ClusPro v2.0 server tool was used to conduct molecular docking for characterizing contact between the illustrated refined vaccine and TLR4 (PDB ID: 4G8A). This server provided 30 different docked models, the best 10 of which were analyzed for the selection of the appropriate vaccine-receptor complex (Supplementary Table 6, Extended data)23. The model showing a strong interaction between vaccine residues and the active site of TLR4 along with the lowest energy value was selected. Model 7 fulfilled the above-mentioned criteria, and thus, was picked as the suitable vaccine-receptor complex (Figure 10). Model 7 had an energy score of -809.1 and indicated the highest binding affinity as it had the lowest energy score (Supplementary Table 6, Extended data)23.

Vaccine surface is represented in yellow, while the receptor is represented in cyan.
To understand the vaccine-receptor compound stability and its large-scale mobility, NMA was carried out using iMODS tool, which is simulated based on intramural configuration of the whole compound. Deformability of the vaccine-immune receptor complex depends on the isolated movement of every amino acid residue, illustrated via chain hinges (Figure 11). The eigenvalue was 2.443455e-557 (Figure 11A). The covariance matrix demonstrates the linkage between duplets of amino acids, the correlated residues marked in red, anti-related residues in white and non-correlated residues in blue, interspersed in dynamical zones (Figure 11B). The elastic mesh-work paradigm classifies which pairs of atoms are interlinked by springs and is visualized as a linking matrix. Dots are colored in accordance to their rigidity; the higher the rigidity, the darker the color (Figure 11C)70.

Molecular dynamic simulation of vaccine-receptor complex, showing (A) eigenvalue; (B) covariance matrix; and (C) elastic network analysis.
The simulated immune response showed resemblance to an actual immune response against a pathogen (Figure 12). Subsequent exposures produced a higher tier immune response compared to the primary immune response. Secondary and tertiary responses showed higher levels of antibodies (i.e., IgM, IgG1, IgG2), which coincides with the waning of antigen (Figure 12A). This demonstrates the development of immune memory cells and, as a result, intensified antigen neutralization upon successive exposure (Figure 12A). Additionally, several B-lymphocyte cell isotypes with prolonged life have been noticed, signifying potential class switching and memory B cell generation (Figure 12B–12C). Increased responses were also observed in helper T cells and cytotoxic T cells with memory generation (Figure 12D–12F). An increase in macrophage activity and engagement was perceived, with a vigorous proliferation of dendritic cells (Figure 12G–12H). An increased amount of IFN-γ and IL-2 cytokines were also noticed (Figure 12l). These findings suggest the development of immune memory and, therefore, immunity against the virus.

In-silico immune response for vaccine construction as an antigen using C-IMMSIM server tool: (A) production of immunoglobulin due to injection of the vaccine; (B) B cell count following consecutive three injections; (C) population of B cell number per state; (D) helper T cell activation; (E) population of helper T cell number per state; (F) population of cytotoxic T cell number per state; (G) per state macrophage population; (H) dendritic cell population; and (I) level of cytokine and interleukins.
RVFV causes higher mortality and fatality among animals than in humans. Since this is a zoonotic virus, it can be transmitted through mosquitos and many regions like Africa, the Arabian Peninsula and their neighbors are at risk of an endemic outbreak7. Thus, a prophylactic measure against RVFV is essential. Vaccination is an ideal approach to gain immunity against infectious agents like RVFV. Development and manufacturing vaccines using a live or attenuated virus takes a huge amount of time and money. Moreover, higher antigenic presence in a weakened vaccine may overstimulate the protective immune system and complicate the situation by causing hypersensitivity reactions21. In comparison with conventional vaccine strategies, multiepitope vaccines possess no such complications71. Epitope-based vaccines could be a great choice in terms of safety, viability and economic rationality. In addition, potency as well as immune responses of multiepitope vaccines can be enhanced through the deliberate engineering of targeted epitopes72. Researchers have been looking for a way to reduce the budget for vaccine development, as well as minimizing allergenicity and side-effects of vaccines for quite a long time. With the emergence of new computational technology and widely available databases, different strategies are now available for designing and developing epitope-based vaccines following immunoinformatics expedients73,74. Vaccines against multitudes of viruses such as SARS-CoV-2, Ebola, Lassa, hepatitis C, Oropouche, Dengue and many more in the pipeline are prime examples of a structural vaccinology approach applied to the design of a multiepitope vaccine model17,19,75–79. In this study, our primary focus was to devise a multiepitope vaccine with the ability to produce robust immunity against RVFV having considered all the parameters of a subunit vaccine.
After retrieving the 232 complete proteomes of RVFV from the ViPR server, screening for antigenic proteins was executed and glycoprotein sequences were selected based on higher antigenicity. A potent multiepitope vaccine should possess the potential to trigger B and T cell activation71. Therefore, potential CTL, HTL as well as LBL epitopic regions of RVFV glycoprotein were chosen for a candidate vaccine modeling. We were particularly interested in integrating B cell epitopes due to its function in antibody production and memory cell formation80. T cell-mediated immunity was also a concern in our vaccine design since plasma cells that give rise to humoral response reactions can easily be saturated by the deluge of antigens. Moreover, lifelong resistance can also be achieved by cell-mediated immunity or cytotoxic T cells81. Cytotoxic T cells can provide lifelong immunity through identifying and destroying infected cells82. Helper T cells, on the other hand, stimulate the release of IL-10, IL-4 and IFN-γ to overcome pro-inflammatory responses and lessen the damage caused to tissues and cells. Besides, they help to produce IgG antibodies and neutralize RVFV infection from peripheral system83. Furthermore, CD4+ cell activate B cells, macrophages, CD8+ cells when they come into contact with an antigen. Thus, all these points were considered and examined during our pursuit of designing the RVFV vaccine.
In a previous study, Adhikari and Rahman predicted overlapping immunodominant T cell epitopes from both nucleocapsid and glycoprotein84. However, their study concluded without formulating the vaccine candidate. Therefore, while Adhikari and Rahman focused on finding overlapping conserved CD8+ and CD4+ epitopes, our work was more centered on selecting CD8+ and CD4+ epitopes with high antigenicity, immunogenicity, non-allergenicity, non-toxicity and eventually tailoring them into a rational and potent vaccine candidate. Herein, unique epitopes from helper and cytotoxic T cells and B cells were chosen not only based on their antigenicity but also on other factors including allergenicity, immunogenicity and toxicity. The complete vaccine design was assembled by attaching the chosen CTL, followed by HTL and finally B cell peptides with the help of AAY, GPGPG, and KK linkers, respectively. Linkers were incorporated in vaccine construction as a part of an essential element to enhance stability, folding, and expression patterns of our vaccine protein85. The adjuvant L7/L12 ribosomal protein was attached to the first CTL epitope using the linker EAAAK. Multiepitope vaccines are less immunogenic when used alone due to a reduction in molecular weight compared to the protein, hence, it requires adjuvant to enhance its efficiency86. Adjuvants are components that help heighten cellular and humoral immunogenic responses for particular antigens as well as amplify the vaccine’s stability and longevity87,88.
Due to the inclusion of immune dominant epitopes, the vaccine candidate was found to have higher antigenicity and immunogenicity while being devoid of allergenic feature. These are the prime features for a vaccine to be effective immune modulator in the first place. When it comes to peptide or subunit vaccines, the size of the vaccine candidate becomes an important matter. Interestingly, our designed multiepitope vaccine is only ~28 kD in size which makes it a suitable candidate vaccine. Solubility is another vital characteristic for any recombinant vaccine16. Luckily, our vaccine construct was predicted to be highly soluble inside the host E. coli system. Furthermore, physicochemical properties of the vaccine candidate were also in suitable range. For instance, the instability index suggested that the chimeric protein would remain stable after synthesis, while the GRAVY value and aliphatic index portrayed the vaccine to be hydrophilic and thermostable, respectively. However, the theoretical pI anticipated our vaccine as acidic which can be adjusted by modifying or adding some basic linkers or additional adjuvants.
The arrangement of crucial protein components was determined by 3D structure modeling, which was used for studying protein dynamics, functions, networking between residues and ligand interactions. The vaccine candidate that showed the best physio-chemical property was chosen and the vaccine construct was refined to significantly improve its expedient properties. The Ramachandran plot revealed 96.6% residues resided within the combined favored and acceptable regions along with only a couple of residues in disallowed region, which confirms the merit of the model. The GDT-HA and MolProbity score, along with clashscore, RMSD value and poor rotamer values were also indicative of sufficiency of the designed vaccine. The Z-score (-6.2) and Verify3D score (80.92%) also signal the satisfactory quality of the vaccine.
Molecular docking as well as molecular dynamics simulation of the TLR4 and vaccine complex were performed in order to gain knowledge about the binding strength, contact nature and structural integrity. The vaccine-receptor complex underwent energy minimization, resulting in boosting of the stability of the total combination. The eigenvalue suggests the stiffness of motion of the molecules within the system and energy needed for their distortion. In our study, eigenvalue increases with each mode passed by which indicates more rigid and compact structure given the time.
Serological evaluation to verify immuno-reactivity can be carried out for validating the candidate vaccine89. Thus, observation of the designed vaccine protein’s expression inside a suitable host is necessary. E. coli has been considered to be a great host choice for determining a recombinant protein’s expression patterns90,91. Hence, codon adaptation was accomplished for our recombinant vaccine to generate high levels of protein expression in E. coli K12. A CAI value of 1.0 and 49.75% GC content suggests the capability of optimum protein synthesis in the host. Moreover, two disulfide bridges were engineered into the vaccine for better stabilization, which is paramount for various biological and biotechnological applications. Animals immunized with glycoproteins of RVFV have been seen to produce protective antibodies against RVFV. Schmaljohn et al. have stated that mice inoculated with Sf9 cells expressing G1 and G2 glycoproteins of RVFV have produced antibodies that are adaptive in nature and neutralizes RVFV infection92. In a study conducted by Faburay et al. construction of a peptide vaccine consisting of two glycoprotein subunits Gn and Gc was carried out against RVFV, and injected into sheep to determine its neutralizing effects. Six sheep were immunized with their constructed subunit vaccine with a dose of 50 μg. The vaccine elicited a strong antibody response against RVFV which was confirmed using enzyme-linked immunosorbent assay (ELISA), suggesting recombinant glycoproteins can be used as a good subunit vaccine candidate. Faburay and his team further carried out a plaque reduction neutralization test to check the amount antibodies produced on primary and secondary dosages. Titer of first dose was low, but the second dosage increased the titer over 128014.
In current study, we also carried out the immune simulation of the vaccine candidate in an in silico immune simulator which demonstrated a good pattern of immune responses. As repetitive vaccine doses enhanced immune responses, we administered three doses that produced a variety of B-cell isotypes and T cell-mediated immune reactions with a noticeably significant number of memory B cells having a half-life of several months. Our simulated immune response shows a stronger and active immune response on secondary and tertiary doses, compared to the initial primary dose. IgG and IgM antibodies had an arbitrary titer of 80000 on second dose and 120000 on third dose. Sustained generation of IFN-γ and IL-2 was observed after multiple exposures of the vaccine as a result of increased helper T cell activation and therefore, the vaccine effectively stimulated a humoral immune response to ramp up immunoglobulin production.
RVFV is a pathogenic agent with potential to become more widespread in upcoming times. Multiepitope vaccines provide a modular and tunable approach to vaccine design compared to traditional vaccine efforts. Our study focused on designing an effective and potent multiepitope vaccine to provide immunity against RVFV. The vaccine contained in silico assessed and predicted CTL, HTL, and LBL epitopes to produce an effective cellular and humoral immunologic response. Moreover, the vaccine simulation showed a potentially protective immune response and passed all molecular simulations with favorable results. However, further validation is required in in vivo and in vitro systems to guarantee the effectiveness of our designed vaccine.
All data underlying the results are available as part of the article and no additional source data are required.
Harvard Dataverse: Extended DATA: Discovery of potential immune epitopes and peptide vaccine design - a prophylactic strategy against Rift Valley Fever Virus. https://doi.org/10.7910/DVN/B9Y1EH23.
This project contains the following extended data in DOCX format:
- Supplementary Table 1 (All initially retrieved complete RVFV protein sequences)
- Supplementary Table 2 (Predicted all potential cytotoxic T-lymphocyte epitopes)
- Supplementary Table 3 (Predicted all potential helper T-lymphocyte epitopes)
- Supplementary Table 4 (Predicted all potential linear B-lymphocyte epitopes)
- Supplementary Table 5 (Physicochemical properties of all designed vaccine candidates)
- Supplementary Table 6 (Scores of top-10 best vaccine-TLR4 docked complexes)
- Supplementary Table 7 (Potential residue pairs for disulfide bridging)
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
Mohammad Minnatul Karim confirms that the author has an appropriate level of expertise to conduct this research, and confirms that the submission is of an acceptable scientific standard. Mohammad Minnatul Karim declares they have no competing interests. Affiliation: Department of Biotechnology and Genetic Engineering, Islamic University, Bangladesh.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)