1Biochemistry and McGill Cancer Centre, McGill University, Montreal, Canada 2Cytokine Signalling Unit, Institut Pasteur, Paris, France
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
The recent development of mammalian target of rapamycin (mTOR) kinase domain inhibitors and genetic dissection of rapamycin-sensitive and -insensitive mTOR protein complexes (mTORC1 and mTORC2) have revealed that phosphorylation of the mTOR substrate 4E-BP1 on amino acids Thr37 and/or Thr46 represents a rapamycin-insensitive activity of mTORC1. Despite numerous previous reports utilizing serine (Ser)-to-alanine (Ala) and threonine (Thr)-to-Ala phosphorylation site mutants of 4E-BP1 to assess which post-translational modification(s) directly regulate binding to eIF4E, an ambiguous understanding persists. This manuscript demonstrates that the initial, rapamycin-insensitive phosphorylation event at Thr46 is sufficient to prevent eIF4E:4E-BP1 binding. This finding is relevant, particularly as mTOR kinase domain inhibitors continue to be assessed for clinical efficacy, since it clarifies a difference between the action of these second-generation mTOR inhibitors and those of rapamycin analogues.
Corresponding author:
Mark Livingstone
Competing interests:
No relevant competing interests declared.
Grant information:
ML was a research student of the Terry Fox Foundation (Award #700029) and is currently supported by the Pasteur Foundation.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The mammalian target of rapamycin (mTOR) protein is an atypical Ser/Thr protein kinase named for its well-characterized inhibition by the natural product rapamycin. Rapamycin-sensitive orthologues of mTOR exist in eukaryotes from yeast to man and are required for growth and proliferation of perhaps all eukaryotic cells. As such, rapamycin has been classified as an anti-fungal agent and is clinically approved as an immunosuppressant and cancer therapy1–3. Well-characterized in vivo substrates of rapamycin-sensitive mTOR activity include the 70 kDa ribosomal protein S6 kinase (S6K1) and the eukaryotic [translation] initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1). The mTOR-dependent phosphorylation site on S6K1, Thr389, is required for kinase activity, explaining rapamycin’s inhibition of S6K1 activity4. 4E-BP1, on the other hand, is subject to multisite phosphorylation culminating in the release of bound eIF4E, leading to an ambiguous understanding of which phosphorylation site(s) regulate(s) eIF4E binding5.
Mammalian 4E-BP1 is subject to an ordered phosphorylation on at least 5 major amino acid residues in response to serum-stimulation, as has been demonstrated by two-dimensional (isoelectric focusing and SDS-PAGE) electrophoresis (2DE)6. This approach of separating post-translationally modified forms of a protein based on charge and apparent molecular weight has proved to be particularly useful when combined with phosphorylation-specific antibodies7. Due to amino acid sequence similarity, phospho-specific anti-Thr37/46 antibodies did not allow determination of whether the initial phosphorylation event is at Thr37 or Thr46 using this technique, although priming phosphorylation at both of these sites is thought to be required for subsequent phosphorylation at Thr70, followed by phosphorylation at an unidentified site, and finally at Ser656,8. Given their positions flanking the amino acids responsible for eIF4E binding (amino acid residues 53–59), it is conceivable that Thr46 and Ser65 are responsible for the phosphorylation-mediated modulation of eIF4E binding occurring in response to mTOR activity. Indeed, a significant body of work supports the role of phosphorylation at Thr46 in regulating eIF4E:4E-BP1 binding9–12. Detailed analyses have led, however, to conflicting results regarding the importance of Ser65 phosphorylation in preventing this protein:protein interaction6,11,13–15.
While rapamycin is effective in blocking phosphorylation at Thr70 and Ser65, phospho-specific antibodies to Thr37/46 show that at least one of these sites is largely rapamycin-insensitive16,17. This residual rapamycin-insensitive phosphorylation is sensitive to serum starvation, amino acid withdrawal, and non-specific phosphatidylinositol 3-kinase (PI3K) and PI3K-like kinase (PIKK) inhibitors16–19. Furthermore, the use of mTOR kinase domain inhibitors (Torin1 and PP242) in combination with mTOR complex 2 (mTORC2)-deficient cells, has allowed the determination that Thr37/46 phosphorylation represents a rapamycin-insensitive function of mTOR complex 1 (mTORC1)20,21.
In vivo studies addressing the relative importance of 4E-BP1 phosphorylation sites have been hampered by its ordered phosphorylation, wherein Thr-to-Ala mutation of Thr37 or Thr46 will block subsequent phosphorylation at Thr70 and Ser65. Mounting circumstantial evidence supports the notion that phosphorylation of Thr37/Thr46 alone is the key event regulating eIF4E:4E-BP1 binding in vivo. Intracellular co-localization of endogenous 4E-BP1 and eIF4E best correlates with dephosphorylation at Thr37/4619. 7-methyl-GTP (cap-column) pull down of eIF4E:4E-BP1 complexes is enhanced by mTOR kinase domain inhibitors more than it is by rapamycin21. Most importantly, however, mTOR active site inhibitors capable of blocking phosphorylation at Thr37/46 (and not rapamycin) induce 4E-BP-dependent phenotypes in cells22.
This manuscript describes new data demonstrating that 4E-BP1 phosphorylation at the initial, mTORC1-dependent, rapamycin-insensitive phosphorylation site is alone in regulating eIF4E binding. Furthermore, this work suggests that Thr46, and not Thr37, is this key phosphorylation site. Given the recent push for pharmaceutical development of kinase inhibitors that block both the rapamycin-sensitive and rapamycin-insensitive activities of mTOR23, a thorough understanding of the importance of rapamycin-insensitive mTORC1 activity is crucial. This manuscript supports the idea that clinically used mTOR kinase domain inhibitors will reduce eIF4E availability much more profoundly than have clinically approved rapamycin analogs.
Materials and methods
Isoelectric focusing combined with SDS-PAGE based two-dimensional electrophoresis was performed as previously described24. Far western analyses were performed as follows using a 32P-labelled eIF4E protein25 with an N-terminal substrate peptide for heart muscle kinase (HMK). One hundred units of bovine HMK was suspended in 10 µl of 40 mM DTT and allowed to stand for 10 minutes. Five micrograms of HMK-eIF4E protein was mixed with 3 µl 10X HMK Buffer (200 mM Tris, pH 7.5, 10 mM DTT, 1 M NaCl, 120 mM MgCl2), 5ul [γ-32P] ATP 3000 Ci/mmol, 1 µl HMK (10U), and water (to 30 µl) and incubated for 45 minutes at 4°C. Probe purification was performed using Pharmacia Nick Column Sephadex G-50 DNA grade. Membranes were subjected to pre-hybridization (25 mM HEPES-KOH, pH 7.7, 25 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.1% NP40, 5% skim milk) for 5 hours at 4°C. Probe hybridization was performed in buffer (20 mM HEPES-KOH, pH 7.7; 75 mM KCl, 2.5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.1% NP40, 1% skim milk) with 250,000 cpm/ml of radiolabelled probe for 10 hours at 4°C. Membranes were washed with hybridization buffer 3 times, 15 minutes prior to exposure to film (BIOMAX MS, Kodak). Following far western analysis, membranes were probed sequentially with antibodies for Phospho-Ser65, Thr70, Thr37/46, and Total 4E-BP1 (Cell Signaling Technology). Cap column based fractionation of cell lysates was performed as previously described26. HeLa S3 and 293 HEK cells were treated with PP242 (2.5 µM, 30 min) or Rapamycin (10 nM, 30 min) unless otherwise indicated. Stable HeLa S3 cell lines expressing wild-type and mutant HA-4E-BP1 proteins were generated using previously described mammalian expression constructs8 and G418 selection. Control siRNA and siRNA for mTOR was from Cell Signaling Technology and delivered using Lipofectamine2000 according to manufacturer’s instructions (Invitrogen). All antibodies were from Cell Signaling Technology and were used according to manufacturer’s instructions. Compounds used were rapamycin (LC Labs), PP242 (Intellikine), torin1 (Tocris), PI-103 (EMD), etoposide and nocodazole (Sigma). Standard laboratory practices were used to control bias and unwanted sources of variability in this study. The primary limitation of the datasets presented in this manuscript is that they represent single biological replicates of an experimental procedure.
Results
4E-BP1 Thr37/46 Phosphorylation is sufficient to block eIF4E binding
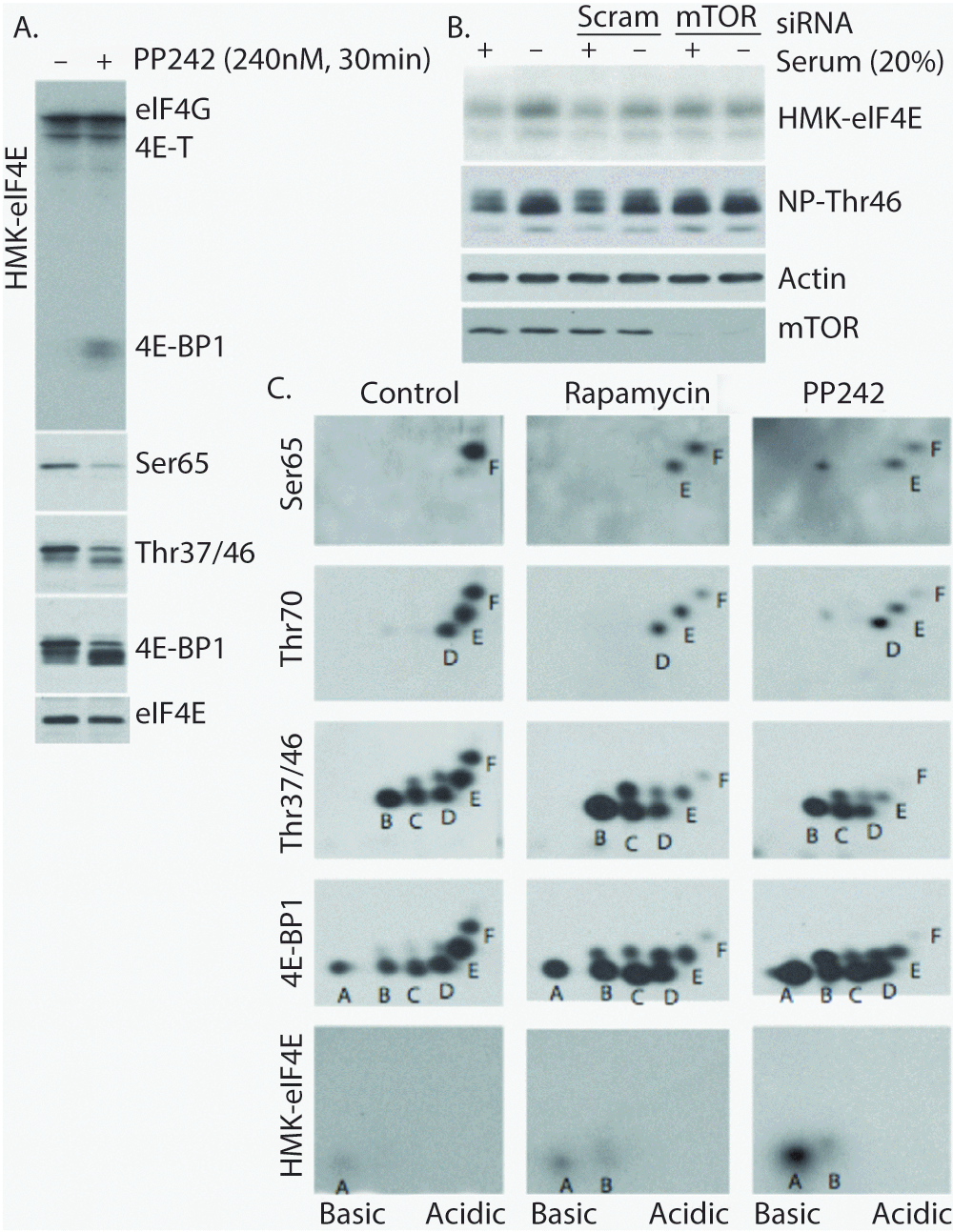
Far western blot analysis, using radiolabeled HMK-eIF4E as a probe, is an effective measure of eIF4E-binding activity11,25. Using this approach, we demonstrate that the treatment of cells with the mTOR kinase inhibitor PP242 results in the following: (i) dramatic increases in the eIF4E-binding competent pool of 4E-BP1, (ii) reduction of 4E-BP1Thr37/46 and Ser65 phosphorylation, and (iii) unaffected binding to bands corresponding in molecular weight to 4E-T and eIF4G (Figure 1A). The mTOR-dependent modulation of eIF4E-binding activity is also apparent under physiological conditions (serum starvation vs. serum stimulation) and can be blocked by siRNA knockdown of mTOR in HeLa S3 cells (Figure 1B). Notably, western blot-based detection of non-phosphorylated (Thr46; denoted NP-Thr46) 4E-BP1 using a rabbit monoclonal antibody (clone: 87D12) recapitulates HMK-eIF4E binding to 4E-BP1.
Figure 1. Analysis of eIF4E binding-competent forms of 4E-BP1.
A) PP242 treatment increases the eIF4E binding ability of 4E-BP1 and reduces 4E-BP1 phosphorylation. B) Physiological treatment (serum starvation) modulates HMK-eIF4E binding ability of 4E-BP1 in an mTOR-dependent manner. Western blot using a non-phospho-4E-BP1/2 (Thr46) antibody parallels eIF4E binding. C) 2DE combined with far-western and western blot analysis to analyze HMK-eIF4E binding forms of 4E-BP1 under control (DMSO) and mTOR inhibitory (Rapamycin and PP242) conditions. Despite fairly equal abundance of spots A-F under control conditions, only "spot A" binds HMK-eIF4E. mTOR inhibition with PP242 potently increases the abundance of eIF4E-binding competent spot A, while rapamycin treatment primarily reduces phosphorylation at Ser65 (spot F).
To more precisely elucidate the molecular modifications of 4E-BP1 induced by PP242 that induce eIF4E binding, whole cell lysates were prepared from HEK293 cells that were subjected to short (30 min) treatment with PP242 or rapamycin followed by 2DE (isoelectric focusing and SDS-PAGE) and western blot analyses (Figure 1C). Under control (DMSO-treated) conditions, the previously described6 hierarchical, multi-site phosphorylation of 4E-BP1 is observed. Here, six differentially phosphorylated forms (labeled A-F) are detected by the total 4E-BP1 antibody, with forms B-F phosphorylated at Thr37 and/or Thr46, D-F phosphorylated at Thr70, and F phosphorylated at Ser65. Far western blot analysis demonstrates that only spot A is competent to bind HMK-eIF4E under control conditions suggesting that the modification responsible for spot B (Thr37 or Thr46) disrupts this interaction. Upon inhibition of mTOR with rapamycin or PP242, the predicted decrease in hyperphosphorylated 4E-BP1 forms is observed with an increase in hypophosphorylated 4E-BP1. While the identity of the phosphorylation event responsible for spot E remains undetermined, these data show that this phosphorylation site is resistant to mTOR inhibition, as rapamycin and PP242-resistant phospho-forms emanating from spot E appear above spots B-D. It is of note that the PP242-induced spot above spot B, which is not phosphorylated at Thr37 or Thr46, represents mono-phosphorylated 4E-BP1 (at the site responsible for spot E) and retains eIF4E-binding ability. The most likely candidates for this site are Thr84, which has shown to be responsible for a similarly slow SDS-PAGE migration11, and Ser101, which has been shown to promote 4E-BP1:Raptor binding27,28. The notion that Ser101 is responsible for spot E is particularly appealing, as this would provide a sound explanation for the hierarchical ordering of this phosphorylation event prior to Ser65 (spot F). That is, it is reasonable to believe that strong 4E-BP1:Raptor binding is required for complete 4E-BP1 phosphorylation, including at Ser65.
4E-BP1 Thr46 is phosphorylated prior to Thr37 under normal growth conditions and prevents association with cap-bound eIF4E
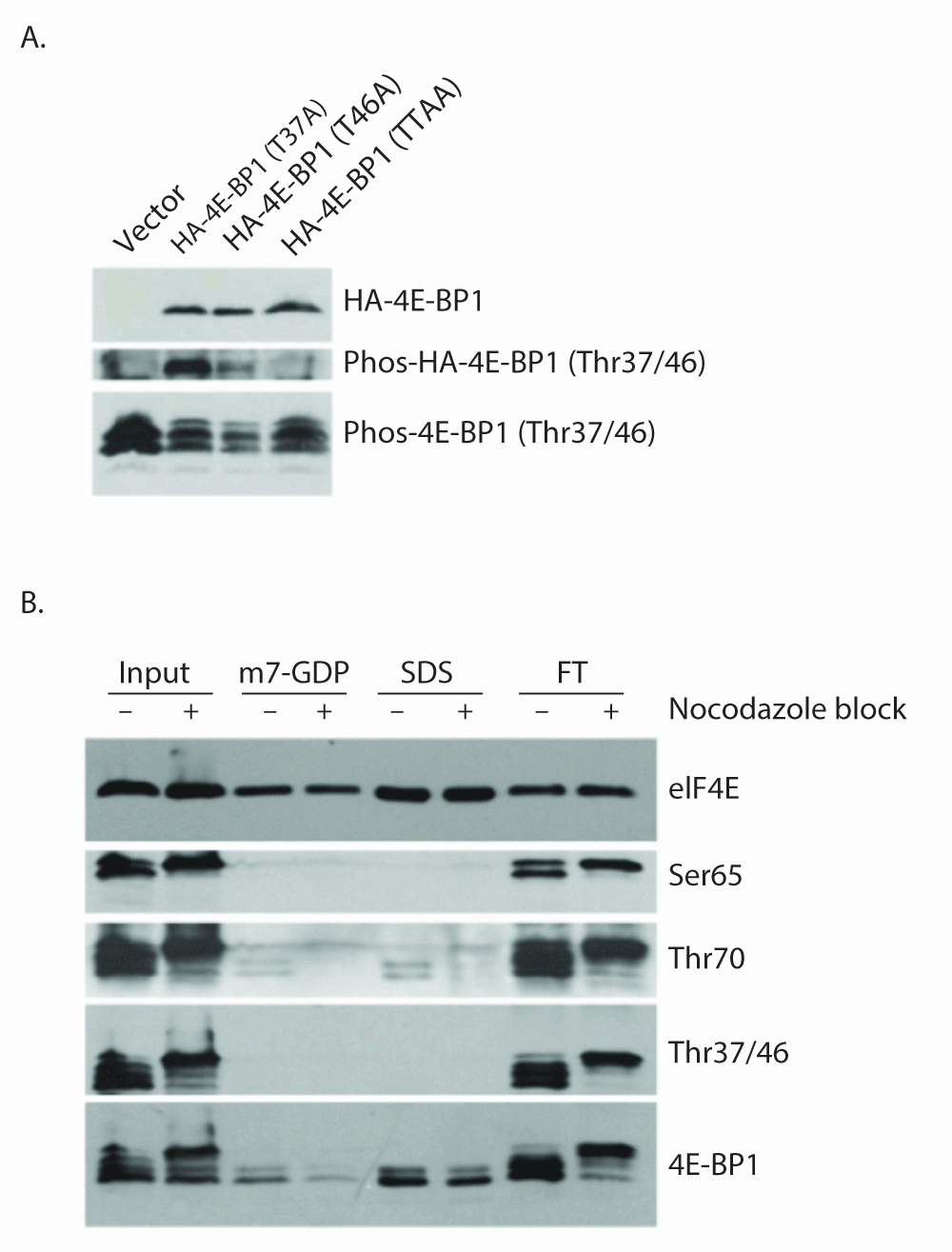
Next, to determine whether Thr37 or Thr46 phosphorylation accounts for "spot B", which is impaired for eIF4E binding, HeLa cell lines stably expressing wild-type or mutant HA-4E-BP1 proteins were generated. Given the ordered phosphorylation of 4E-BP1, mutation of the primary phosphorylation site should block mutation of that of the subsequent phosphorylation site. For this reason, we analyzed Thr37Ala and Thr46Ala mutants in vivo using the phospho-4E-BP1 Thr37/46 antibody to determine whether the preclusion of phosphorylation at one site blocks phosphorylation at the other (Figure 2A). While endogenous 4E-BP1 was detected by the phospho-4E-BP1 Thr37/46 antibody in lysates from all stably selected cell lines, exogenous HA-tagged 4E-BP1 was poorly detected in the Thr46Ala mutant sample, suggesting that Thr46 phosphorylation is required for subsequent Thr37 phosphorylation under normal growth conditions. These results indicate that Thr46 is the initial phosphorylation site responsible for the shift from spot A to spot B, thus phosphorylation at this site alone may be sufficient to prevent 4E-BP1 binding to eIF4E (in Figure 1C). This conclusion, that Thr46 phosphorylation precedes Thr37 phosphorylation, has previously been reached by another group29.
Figure 2. Thr46 phosphorylation is required for Thr37 phosphorylation and is sufficient to prevent eIF4E:4E-BP1 binding.
A) HeLa S3 cells stably expressing 4E-BP1 mutant proteins were subjected to western blotting using anti-HA antibody (upper), and phospho-4E-BP1 (Thr37/46) antibody (middle and lower panels). While phospho-4E-BP1 (Thr37/46) antibodies fail to detect the Thr46Ala single and Thr37/46Ala double point mutant proteins, the Thr37Ala protein is still recognized suggesting that Thr37 is not required for Thr46 phosphorylation. Endogenous 4E-BP1 phosphorylated at Thr37/46 is shown as a control. B) Untreated and Nocodazole-blocked HeLa S3 cells were subjected to cap-column pull down of eIF4E and associated proteins. eIF4E was eluted with m7-GDP followed by SDS, and fractions were analyzed by western blot using eIF4E and 4E-BP1 antibodies and phospho-4E-BP1 antibodies.
This model, wherein 4E-BP1 phosphorylation at the initial phosphorylation site (Thr46) is sufficient to prevent eIF4E binding, is also supported by 7-methyl-GTP (cap-column) pull down data (Figure 2B). Here the cap-column serves as a molecular mimic for the mRNA 5´-cap allowing eIF4E and associated binding proteins to be isolated from cell lysates. As a chemically induced pseudo-mitotic state has previously been shown to dramatically modulate the phosphorylation of 4E-BP126,30, nocodazole treatment was employed to potentially increase the diversity of 4E-BP1 phospho-forms present within our lysates. Control and nocadazole-blocked HeLa S3 cells were subjected to cap-column pull-down of eIF4E and associated 4E-BP1. This technique allowed detectable binding of only the fastest SDS-PAGE migrating forms of 4E-BP1, indicating differential binding between hypophosphorylated and hyperphosphorylated 4E-BP1 had occurred. The use of phospho-specific antibodies demonstrates that Thr37/Thr46 phosphorylated 4E-BP1 is not detectably present in this cap-column bound eIF4E fraction. Trace amounts of Thr70 phosphorylated 4E-BP1 are detected in these lanes, suggesting that mono-phosphorylated (at Thr70) 4E-BP1 exists and is eIF4E binding competent. A similar conclusion was recently reached by another group31. It should be noted that the total 4E-BP1 antibody detects a doublet band in the cap-column bound eIF4E fraction. The upper band of this doublet likely represents 4E-BP1 phosphorylated at the site responsible for the above-described "spot E"; a phospho-form which retains eIF4E-binding ability.
Existence of alternative 4E-BP1 phosphorylation patterns
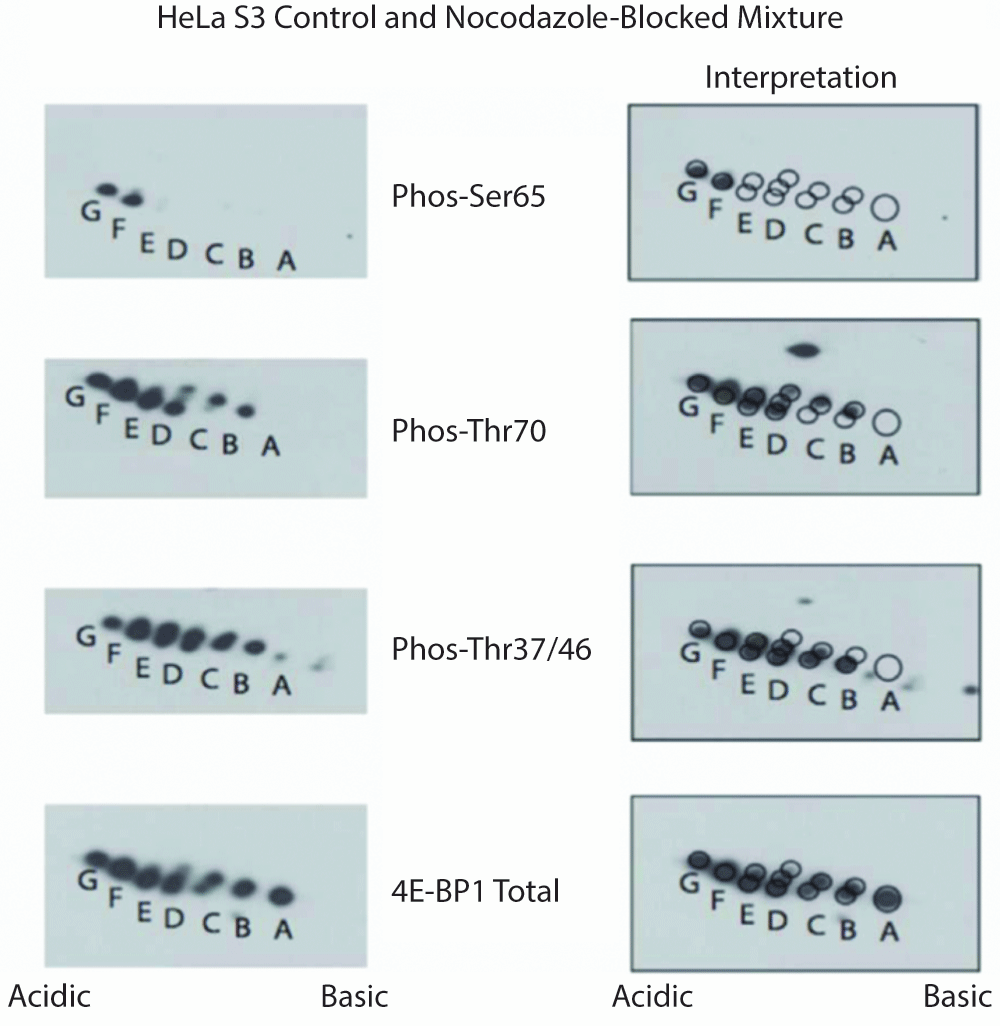
To further explore the potential existence of 4E-BP1 phospho-forms failing to adhere to the strict hierarchical phosphorylation pattern Thr37/46->Thr70->spot E->Ser65, untreated and nocodazole-blocked HeLa S3 cell lysates were mixed and subjected to 2D-E. Nocodazole block was again used to induce aberrant phosphorylation patterns of 4E-BP1. As shown in Figure 3, a pattern that was distinct from that produced under normal growth conditions emerged. Here, in addition to the normal hierarchical phosphorylation described above, alternative mono-phosphorylated species were present, as was an additional phosphorylation site causing the appearance of "spot G". Although, this chemical-induced atypical phosphorylation pattern may not exist under physiological conditions, these data provide support for the existence of these alternative 4E-BP1 phospho-forms.
Figure 3. 4E-BP1 phosphorylation is not strictly ordered.
Untreated and nocodazole-blocked HeLa S3 cells lysed, pooled and subjected to 2D-E prior to analysis with phospho-specific and total 4E-BP1 antibodies (left panels). In addition to the standard ordered phosphorylation (Thr37 or Thr46, then "spot B", then Thr70, then "spot E", then Ser65, then "spot G"), 4E-BP1 singly phosphorylated at Thr70 is also observed indicating that this species can exist in vivo. To facilitate interpretation, the same images have been overlaid with a grid of circles corresponding to spots visible with the total 4E-BP1 antibody (right panels).
PP242 inhibits etoposide-induced phosphorylation of Ser/Thr-Gln motifs
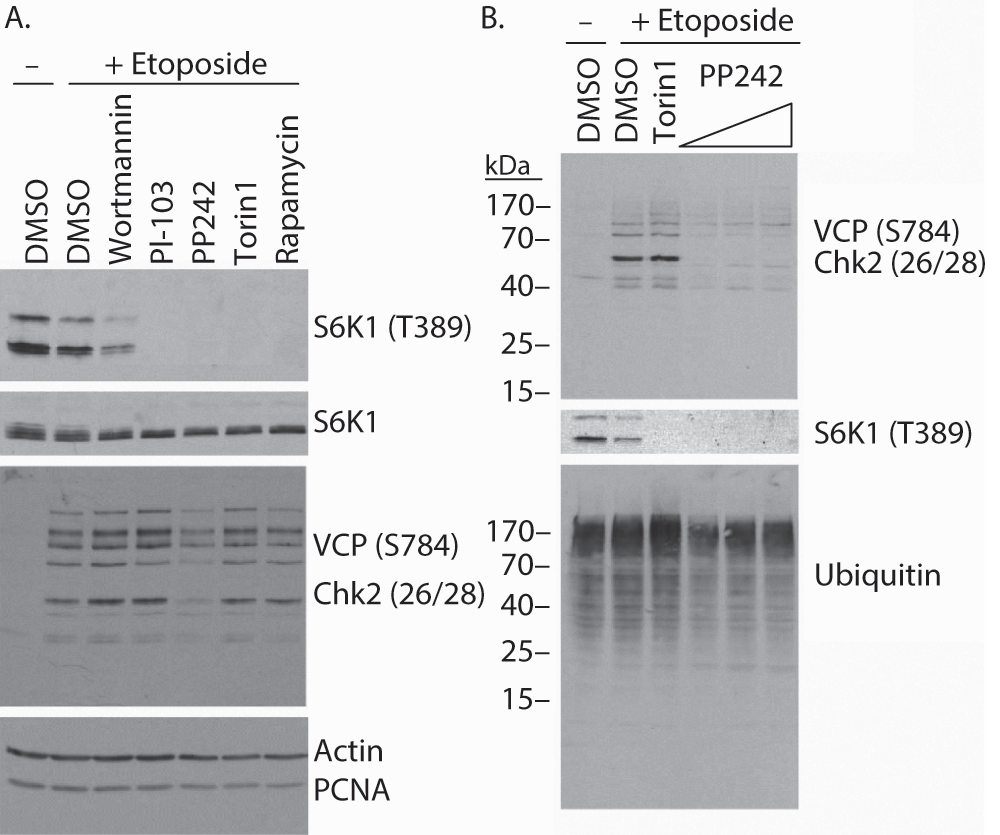
Recently it has been reported that among mTOR kinase domain inhibitors, PP242 exhibits remarkably low specificity compared with Torin1, KU63794 and WYE35432. Notably, both PP242 and Torin1 were shown to bind multiple PI3K-related kinases (PIKKs), including three key DNA damage-activated kinases ATM, ATR and DNAPK, although cell-based assays failed to show inhibition of these kinases. To assess the specificity of PP242 in our system, we employed a broadly reactive phospho-specific antibody capable of recognizing multiple phospho-Ser/Thr-Gln substrates of ATM, ATR and DNAPK33,34. Indeed, with this antibody, we observed multiple etoposide-induced bands by western blot analysis (Figure 4), including bands corresponding to VCP phosphorylated at Ser784 (97 kDa) and Chk2 phosphorylated at Thr26/Ser28 (62 kDa). Remarkably, of the mTOR/PI3K inhibitors tested, only PP242 visibly reduced the etoposide-induced phosphorylation of these PIKK substrates (Figure 4A). As the concentration of PP242 (2.5 µM) was higher than that used for the other inhibitors (wortmannin 0.1 µM, PI-103 1.0 µM, Torin1 0.25 µM and Rapamycin 0.01 µM), PP242 pretreatment was also assessed at 0.34, 0.68 and 1.25 µM, and gave similar results (Figure 4B).
Figure 4. PP242 inhibits etoposide-induced phosphorylation of Ser/Thr-Gln motifs.
A) Western blot analysis of lysates from HeLa cells pretreated with the indicated inhibitors prior to etoposide treatment reveals that PP242 reduces the appearance of multiple phospho-SQ/TQ epitopes recognized by the broadly reactive phospho-VCP/Chk2 antibody. B) Varying concentrations of PP242 from 0.34–1.25 µM block etoposide-induced phosphorylation of proteins recognized by the phospho-SQ/TQ antibody.
Discussion
Although, our conclusion that phosphorylation at Thr46 is the key event regulating 4E-BP1:eIF4E binding has been suggested previously, none of these previous studies unambiguously established that Thr46 alone is the key important site. These studies either: (i) employed in vivo phosphorylation of a Thr46Ala point mutant also blocking subsequent phosphorylation events9,11; (ii) failed to identify a single important phosphorylation site10,11; or (iii) employed in vitro phosphorylation of Thr46Ala point mutant using a non-physiological kinase11,12.
Importantly, equally credible work has reported that phosphorylation on Thr46 is unimportant in the regulation of 4E-BP1:eIF4E binding8,35. It is interesting to note that both of these studies evaluate the capacity of in vitro phosphorylation at Thr46 to disrupt pre-existing 4E-BP1:eIF4E complexes and utilize cap column purification to isolate eIF4E-bound 4E-BP1. This experimental detail is particularly relevant, as it has been shown that the RNA cap can exert an allosteric effect stabilizing 4E-BP1:eIF4E binding14,36. Taken together, our data and these previous reports could suggest that Thr46 phosphorylation is sufficient to block the initial binding between eIF4E and 4E-BP1 as observed by far western analyses, but that hyperphosphorylation of 4E-BP1, including at Ser65, is required to disrupt existing 4E-BP1:eIF4E complexes.
The far western analysis of 2DE separated 4E-BP1 phospho-forms presented in Figure 1C is apparently at the limit of its useful range of detection. While this technique allows a comparison of binding efficiencies of spots A and B to evaluate the impact of Thr46 phosphorylation, it is not useful to compare the relative binding abilities of spots E and F to allow a similar assessment of the importance of Ser65 phosphorylation. Perhaps with a more sensitive assay we would have observed a similar decrease in eIF4E binding upon Ser65 phosphorylation. This would be evidence that Thr46 phosphorylation is sufficient to block the initial binding of 4E-BP1 to eIF4E when both proteins are present at low physiological concentrations, but that phosphorylation at multiple sites culminating at Ser65 is required to prevent/disrupt binding when the two proteins are present at high concentrations/local concentrations.
The data presented in Figure 2B at first seem to be at odds with this theory that 4E-BP1 phosphorylated at Thr37 or Thr46 could be pre-associated with mRNA 5´ cap-bound eIF4E in cells. This technique assesses, however, the de novo association of eIF4E (4E-BP1 associated or not) with an mRNA 5´ cap analog, therefore any pre-existing complexes must dissociate from cellular mRNA caps prior to isolation. These data do tell us that if there is some pool of 4E-BP1 phosphorylated on Thr46 associated with eIF4E, then this complex is not able to efficiently bind the mRNA cap analog. This assay may also evaluate the sequential binding of eIF4E to the cap followed by 4E-BP1 binding to cap-associated eIF4E and indicate in agreement with the far western data in Figure 1C that de novo binding is blocked by Thr46 phosphorylation.
Figure 2B and Figure 3 demonstrate that blocking cells in a pseudo-mitotic state with nocodazole results in phosphorylation of 4E-BP1 at up to 6 distinct phosphorylation sites, a finding at odds with previous work demonstrating hypophosphorylation of 4E-BP1 in mitosis26. The polyclonal antibody, referred to as 11208 and used in this previous study, however, exhibits selectivity for the non-phosphorylated forms of the protein after nocodazole treatment (ML, unpublished). A similar faulty antibody-based discrepancy regarding the phosphorylation state of 4E-BP1 in meiotic oocytes was resolved37. While we observed at most 6 different phospho-forms of 4E-BP1, the PhosphoSitePlus database38 indicates that 20 distinct phosphorylation sites have been described in the literature and/or mass spectrometry-based datasets. This discrepancy suggests that many of these 20 phosphorylation events are either mutually exclusive, or occur only rarely, not at all, in response to specific stimuli, or in specific cell types.
Finally, the demonstration presented within this manuscript that PP242 exhibits poor specificity for mTOR in a cell-based assay for PIKK off-target effects likely does not change the conclusions of this manuscript; however, it serves as a reminder that any new mTOR inhibitor may have unanticipated effects. While the inhibition of mTORC2 by mTOR kinase domain inhibitors is expected to have profound effects, the evidence presented in this manuscript suggests that the rapamycin-insensitive activity of mTORC1 towards 4E-BP1 will also be quite important as the clinical safety and efficacy of mTOR kinase domain inhibitors is assessed.
Author contributions
ML conceived the study, designed the experiments, and prepared the first draft of the manuscript. ML and MB carried out the research, were involved in the revision of the draft manuscript, and have approved the final content.
Competing interests
No relevant competing interests declared.
Grant information
ML was a research student of the Terry Fox Foundation (Award #700029) and is currently supported by the Pasteur Foundation.
Acknowledgements
We thank Marc R. Fabian for assistance with far western analyses.
Faculty Opinions recommended
References
1.
Vezina C, Kudelski A, Sehgal SN:
Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomyceteand isolation of the active principle.
J Antibiot (Tokyo).
1975; 28(10): 721–726. PubMed Abstract
| Publisher Full Text
2.
Martel RR, Klicius J, Galet S:
Inhibition of the immune response by rapamycin, a new antifungal antibiotic.
Can J Physiol Pharmacol.
1977; 55(1): 48–51. PubMed Abstract
3.
Hudes GR, Berkenblit A, Feingold J, et al.:
Clinical trial experience with temsirolimus in patients with advanced renal cell carcinoma.
Semin Oncol.
2009; 36(Suppl 3): S26–36. PubMed Abstract
| Publisher Full Text
4.
Pearson RB, Dennis PB, Han JW, et al.:
The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain.
EMBO J.
1995; 14(21): 5279–5287. PubMed Abstract
| Free Full Text
5.
Beretta L, Gingras AC, Svitkin YV, et al.:
Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation.
EMBO J.
1996; 15(3): 658–664. PubMed Abstract
| Free Full Text
6.
Gingras AC, Raught B, Gygi SP, et al.:
Hierarchical phosphorylation of the translation inhibitor 4E-BP1.
Genes Dev.
2001; 15(21): 2852–2864. PubMed Abstract
| Free Full Text
7.
Kovarova H, Hajduch M, Livingstone M, et al.:
Analysis of state-specific phosphorylation of proteins by two-dimensional gel electrophoresis approach.
J Chromatogr B Analyt Technol Biomed Life Sci.
2003; 787(1): 53–61. PubMed Abstract
| Publisher Full Text
9.
Burnett PE, Barrow RK, Cohen NA, et al.:
RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1.
Proc Natl Acad Sci USA.
1998; 95(4): 1432–1437. PubMed Abstract
| Publisher Full Text
| Free Full Text
10.
Fadden P, Haystead TA, Lawrence JC Jr:
Identification of phosphorylation sites in the translational regulator, PHAS-I, that are controlled by insulin and rapamycin in rat adipocytes.
J Biol Chem.
1997; 272(15): 10240–10247. PubMed Abstract
| Publisher Full Text
11.
Mothe-Satney I, Yang D, Fadden P, et al.:
Multiple mechanisms control phosphorylation of PHAS-I in five (S/T)P sites that govern translational repression.
Mol Cell Biol.
2000; 20(10): 3558–3567. PubMed Abstract
| Publisher Full Text
| Free Full Text
12.
Yang D, Brunn GJ, Lawrence JC Jr:
Mutational analysis of sites in the translational regulator, PHAS-I, that are selectively phosphorylated by mTOR.
FEBS Lett.
1999; 453(3): 387–390. PubMed Abstract
| Publisher Full Text
13.
Tomoo K, Abiko F, Miyagawa H, et al.:
Effect of N-terminal region of eIF4E and Ser65-phosphorylation of 4E-BP1 on interaction between eIF4E and 4E-BP1 fragment peptide.
J Biochem.
2006; 140(2): 237–246. PubMed Abstract
| Publisher Full Text
14.
Tomoo K, Matsushita Y, Fujisaki H, et al.:
Structural basis for mRNA Cap-Binding regulation of eukaryotic initiation factor 4E by 4E-binding protein, studied by spectroscopic, X-ray crystal structural, and molecular dynamics simulation methods.
Biochim Biophys Acta.
2005; 1753(2): 191–208. PubMed Abstract
| Publisher Full Text
15.
Tait S, Dutta K, Cowburn D, et al.:
Local control of a disorder-order transition in 4E-BP1 underpins regulation of translation via eIF4E.
Proc Natl Acad Sci USA.
2010; 107(41): 17627–17632. PubMed Abstract
| Publisher Full Text
| Free Full Text
16.
Fan QW, Knight ZA, Goldenberg DD, et al.:
A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma.
Cancer Cell.
2006; 9(5): 341–349. PubMed Abstract
| Publisher Full Text
| Free Full Text
17.
Wang X, Beugnet A, Murakami M, et al.:
Distinct signaling events downstream of mTOR cooperate to mediate the effects of amino acids and insulin factor 4E-binding proteins.
Mol Cell Biol.
2005; 25(7): 2558–2572. PubMed Abstract
| Publisher Full Text
| Free Full Text
19.
Livingstone M, Larsson O, Sukarieh R, et al.:
A chemical genetic screen for mTOR pathway inhibitors based on 4E-BP-dependent nuclear accumulation of eIF4E.
Chem Biol.
2009; 16(12): 1240–1249. PubMed Abstract
| Publisher Full Text
20.
Feldman ME, Apsel B, Uotila A, et al.:
Active-Site Inhibitors of mTOR Target Rapamycin-Resistant Outputs of mTORC1 and mTORC2.
PLoS Biol.
2009; 7(2): e38. PubMed Abstract
| Publisher Full Text
| Free Full Text
21.
Thoreen CC, Kang SA, Chang JW, et al.:
An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1.
J Biol Chem.
2009; 284(12): 8023–8032. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Dowling RJ, Topisirovic I, Alain T, et al.:
mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs.
Science.
2010; 328(5982): 1172–1176. PubMed Abstract
| Publisher Full Text
| Free Full Text
23.
Benjamin D, Colombi M, Moroni C, et al.:
Rapamycin passes the torch: a new generation of mTOR inhibitors.
Nat Rev Drug Discov.
2011; 10(11): 868–880. PubMed Abstract
| Publisher Full Text
24.
Bidinosti M, Ran I, Sanchez-Carbente MR, et al.:
Postnatal deamidation of 4E-BP2 in brain enhances its association with raptor and alters kinetics of excitatory synaptic transmission.
Mol Cell.
2010; 37(6): 797–808. PubMed Abstract
| Publisher Full Text
| Free Full Text
25.
Pause A, Belsham GJ, Gingras AC, et al.:
Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5´-cap function.
Nature.
1994; 371(6500): 762–767. PubMed Abstract
| Publisher Full Text
27.
Beugnet A, Wang X, Proud CG:
Target of rapamycin (TOR)-signaling and RAIP motifs play distinct roles in the mammalian TOR-dependent phosphorylation of initiation factor 4E-binding protein 1.
J Biol Chem.
2003; 278(42): 40717–40722. PubMed Abstract
| Publisher Full Text
28.
Wang X, Li W, Parra JL, et al.:
The C terminus of initiation factor 4E-binding protein 1 contains multiple regulatory features that influence its function and phosphorylation.
Mol Cell Biol.
2003; 23(5): 1546–1557. PubMed Abstract
| Publisher Full Text
| Free Full Text
29.
Herbert TP, Tee AR, Proud CG:
The extracellular signal-regulated kinase pathway regulates the phosphorylation of 4E-BP1 at multiple sites.
J Biol Chem.
2002; 277(13): 11591–11596. PubMed Abstract
| Publisher Full Text
30.
Heesom KJ, Gampel A, Mellor H, et al.:
Cell cycle-dependent phosphorylation of the translational repressor eIF-4E binding protein-1 (4E-BP1).
Curr Biol.
2001; 11(17): 1374–1379. PubMed Abstract
| Publisher Full Text
31.
Ayuso MI, Hernandez-Jimenez M, Martin ME, et al.:
New hierarchical phosphorylation pathway of the translational repressor eIF4E-binding protein 1 (4E-BP1) in ischemia-reperfusion stress.
J Biol Chem.
2010; 285(45): 34355–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
32.
Liu Q, Kirubakaran S, Hur W, et al.:
Kinome-wide selectivity profiling of ATP-competitive mammalian target of rapamycin (mTOR) inhibitors and characterization of their binding kinetics.
J Biol Chem.
2012; 287(13): 9742–9752. PubMed Abstract
| Publisher Full Text
| Free Full Text
33.
Livingstone M, Ruan H, Weiner J, et al.:
Valosin-containing protein phosphorylation at Ser784 in response to DNA damage.
Cancer Res.
2005; 65(17): 7533–7540. PubMed Abstract
35.
Heesom KJ, Denton RM:
Dissociation of the eukaryotic initiation factor-4E/4E-BP1 complex involves phosphorylation of 4E-BP1 by an mTOR-associated kinase.
FEBS Lett.
1999; 457(3): 489–493. PubMed Abstract
| Publisher Full Text
36.
Shen X, Tomoo K, Uchiyama S, et al.:
Structural and thermodynamic behavior of eukaryotic initiation factor 4E in supramolecular formation with 4E-binding protein 1 and mRNA cap analogue, studied by spectroscopic methods.
Chem Pharm Bull (Tokyo).
2001; 49(10): 1299–1303. PubMed Abstract
| Publisher Full Text
37.
Ellederova Z, Kovarova H, Melo-Sterza F, et al.:
Supression of translation during in vitro maturation of pig oocytes despite enhanced formation of cap-binding protein complex eIF4F and 4E-BP1 hyperphosphorylation.
Mol Reprod Dev.
2006; 73(1): 68–76. PubMed Abstract
| Publisher Full Text
38.
Hornbeck PV, Kornhauser JM, Tkachev S, et al.:
PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse.
Nucleic Acids Res.
2012; 40(Database issue): D261–270. PubMed Abstract
| Publisher Full Text
| Free Full Text
Comments on this articleComments (1)
Version 1
VERSION 1PUBLISHED 18 Jul 2012
Reader Comment
20 Jul 2012
Juming Jan, State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, China
20 Jul 2012
Reader Comment
The paper clearly elucidate the functionally important role of the phosphorylated Thr46 of 4EBP1 in the course of 4EBP1 binding with eIF4E. the design of experiments is very persuading, especially
...
Continue readingThe paper clearly elucidate the functionally important role of the phosphorylated Thr46 of 4EBP1 in the course of 4EBP1 binding with eIF4E. the design of experiments is very persuading, especially the 4EBP1 point mutant and pull down of eIF4E give a strong proof to unravel the crucial role of Thr46 phosphorylation in binding eIF4E. Thank the authors for providing the powerful evidences to make me under the mechanism of the interaction between 4EBP1 and eIF4E.
The eIF4E are released from the 4EBP1 in many malignant tumor due to the hyperphosphorylation of 4EBP1. It will be very helpful to discover some new mTOR signal inhibitors and to explain the mechanism, if the processes of 4EBP1 dephosphorylation are clarified.
The paper clearly elucidate the functionally important role of the phosphorylated Thr46 of 4EBP1 in the course of 4EBP1 binding with eIF4E. the design of experiments is very persuading, especially the 4EBP1 point mutant and pull down of eIF4E give a strong proof to unravel the crucial role of Thr46 phosphorylation in binding eIF4E. Thank the authors for providing the powerful evidences to make me under the mechanism of the interaction between 4EBP1 and eIF4E.
The eIF4E are released from the 4EBP1 in many malignant tumor due to the hyperphosphorylation of 4EBP1. It will be very helpful to discover some new mTOR signal inhibitors and to explain the mechanism, if the processes of 4EBP1 dephosphorylation are clarified.
Competing Interests:No competing interests were disclosed.Close
ML was a research student of the Terry Fox Foundation (Award #700029) and is currently supported by the Pasteur Foundation.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Livingstone M and Bidinosti M. Rapamycin-insensitive mTORC1 activity controls eIF4E:4E-BP1 binding [version 1; peer review: 3 approved]. F1000Research 2012, 1:4 (https://doi.org/10.12688/f1000research.1-4.v1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
This submission is quite nice for experts, but not particularly well-written for those outside of the field of 4E-BP1 phosphorylation, who will have difficulty reading it ; hence my suggested clarifications below. The article is important as it demonstrates that
... Continue reading
This submission is quite nice for experts, but not particularly well-written for those outside of the field of 4E-BP1 phosphorylation, who will have difficulty reading it ; hence my suggested clarifications below. The article is important as it demonstrates that the use of mTOR-kinase domain inhibitors would be more efficacious than the use of rapamycin analogs as therapeutics.
The work clarifies that phosphorylation of 4E-BP1 at Ser65 by mTOR is not required to inhibit 4E-BP1 binding to eIF4E and that rapamycin-insensitive mTOR-mediated phosphorylation at 4E-BP1 Thr46 is sufficient to inhibit 4E-BP1 binding to eIF4E. Phosphorylation at 4E-BP1 Thr37 is also shown to be rapamycin-insensitive. Notably, evidence that phosphorylation at Thr 37 or Thr 46 appears to be required for phosphorylation at Thr 70 and Ser 65 supported the notion that the earlier phosphorylation events were key to promoting translation.
Improvement Suggestions:
Page 2, first paragraph, last sentence. The meaning of “4E-BP-dependent phenotypes” is not particularly clear.
Page 2, line four of Materials and Methods. Shouldn’t “32″ be superscript?
Page 2, last paragraph. The statement “Here, six differentially phosphorylated forms (A-F) …” is confusing. Is form A really phosphorylated? Earlier in the manuscript, the authors mentioned 5 major phosphorylation sites.
In Figure 1, the assay used in this Figure requires much better description in the text. Part A of the legend requires specification of the experimental approach. Also, the title of Figure 1 could be more useful if it were informative/conclusive rather than descriptive.
Page 3, right column, second paragraph. The sentence “While endogenous… , suggesting that Thr46 phosphorylation is required for subsequent The37 phosphorylation under normal growth conditions.” is misleading since 4E-BP1 mutant (Thr46Ala) is clearly phosphorylated at Thr37 in Fig. 2A middle panel. With the data presented here the reader is left wondering whether the differences in detecting HA-4E-BP1 (T37A) or HA-4E-BP1 (T46A) might be simply due to different affinities of the (The37/46) antibody used.
What is the difference between the middle and the lower panel? Are the antibodies used to probe the middle and the lower panel (e.g. phosho-HA-4E-BP1 (Thr37/46) vs. phosho-4E-BP1 (Thr37/46)) different or the same? When using cell lysates of stable cell lines and a phospho-4E-BP1 (Thr37/46) antibody, shouldn’t be HA 4E-BP1 wildtype and mutant forms be detectable in the lower panel as well?
Page 3, last paragraph, line 8. “modulate” would be clearer if described as the specific modulations that occur – in the literature a state of 4E-BP1 hypophosphorylation. The author understands that the changes are complicated and clarified from these authors’ perspective in Figure 3 and in the Discussion. However, possibly, the reader could be better informed at this point of the manuscript.
Page 4, second paragraph, right-hand column. Please clarify the effects and usefulness of etoposide and the meaning of VCP and, in the legend, VCP/Chk2.
In Figure 2B, the authors should provide a control western blot of an unrelated protein which does not belong to the eIF4E-associated proteins under normal conditions to show the unspecific background of this assay.It is not clear to this reader why mono-phosphorylated 4E-BP1 (at position Thr70) is detectable in the non-Nocadazole treated cell samples in Fig. 2B (lane 3 and 5) when in Fig.3 they show that mono-phosphorylation of 4E-BP1 at position Thr70 is triggered by a Nocodazol-induced block of Hela cells but not detectable under normal conditions.
In Figure 3, why did the authors mix Control and Nocodazole-blocked Hela cell lysates instead of showing them side-by-side? Also, why are there additional spots in the interpretation panels for Phos-Thr70 and Phos-Thr37/46 that have no counterpart in the respective western blot panels? This figure is already difficult, so the authors should be careful with the overlays.
In Figure 4, all results in this Figure are not adequately described. For example, the authors need to explain the analysis of ubiquitin and its significance to their studies.
The sentence “ …rapamycin-insensitive phosphorylation site is alone in regulating eIF4E binding.” should read something like “ …rapamycin-insensitive phosphorylation site is alone responsible in regulating eIF4E binding”.
Competing Interests: No competing interests were disclosed.
We confirm that we have read this submission and believe that we have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Through binding to eIF4E, the 4EBP proteins (4EBP1, 2, 3) interfere with formation of the eIF4F cap-binding complex. A key function of the mTOR serine-threonine kinase is to phosphorylate 4EBPs on multiple sites, which blocks their ability to bind to
... Continue reading
Through binding to eIF4E, the 4EBP proteins (4EBP1, 2, 3) interfere with formation of the eIF4F cap-binding complex. A key function of the mTOR serine-threonine kinase is to phosphorylate 4EBPs on multiple sites, which blocks their ability to bind to eIF4E thus potentiating cap-dependent translation.
Recently it has become clear that ATP-competitive mTOR kinase inhibitors are more effective than the allosteric compound rapamycin at blocking mTOR-dependent 4EBP phosphorylation, particularly on sites T37 and T46. This greater effect of mTOR kinase inhibitors on 4EBP phosphorylation correlates with increased anti-proliferative and pro-apoptotic effects. The purpose of the current study is to determine the order of phosphorylation of sites T37 and T46, and which site is most important for regulating binding of 4EBP1 to eIF4E. This is a fairly narrow question to address but the authors argue that it is important for understanding drug mechanism and that previous approaches have not addressed this question adequately.
Overall the data support the conclusion that T46 phosphorylation is the initial, rapamycin-insensitive event that blocks 4EBP1-eIF4E binding. The main approach used is two-dimensional electrophoresis (IEF followed by SDS-PAGE), then far-western blotting with labeled eIF4E to determine which spots are capable of eIF4E binding. Figure 1A provides a 1-D blot that shows convincingly that the far-western technique can reveal increased binding of eIF4E to 4EBP1 in cells treated with the mTOR kinase inhibitor PP242. Figure 1B shows that 4EBP1 phosphorylation and release from eIF4E are induced by serum in a mTOR-dependent manner. Figure 1C presents the 2D data illustrating hierarchical phosphorylation and distinct effects of rapamycin and PP242. The data support the conclusion that only a small subset of 4EBP forms are capable of binding eIF4E, that these forms lack T37/46 phosphorylation, and that PP242 increases this pool to a greater extent than rapamycin. To distinguish the roles of T37 and T46 they express HA-tagged 4EBP1 and alanine substitution mutants in HeLa cells. The data in Figure 2A show that T46A mutation, but not T37A mutation, blocks detection by the phospho-T37/46 antibody. This suggests that T46 phosphorylation precedes and is required for T37 phosphorylation. A cap binding assay in Figure 2B supports the conclusion that T37/46-phosphorylated 4EBP1 cannot bind eIF4E in the cap-binding complex, but this is not novel and the technique does not add further evidence for initial T46 phosphorylation.
The rest of the data seem added on and superfluous to the main message of the paper. The experiment in Figure 3 shows 2D far western blotting data on lysates of nocodazole-treated cells, which artificially increases the diversity of 4EBP1 spots. As the authors point out, these might not exist under physiological conditions so the relevance is unclear. Figure 4 addresses an entirely different problem, namely the lack of selectivity of the PP242 compound. While it is of some interest that PP242 differs from other compounds of this class in its apparent inhibition of DNA-PK and related kinases, this observation is too peripheral to the main study and should be developed further before publication.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Juming Jan, State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, China
20 Jul 2012
Reader Comment
The paper clearly elucidate the functionally important role of the phosphorylated Thr46 of 4EBP1 in the course of 4EBP1 binding with eIF4E. the design of experiments is very persuading, especially
...
Continue readingThe paper clearly elucidate the functionally important role of the phosphorylated Thr46 of 4EBP1 in the course of 4EBP1 binding with eIF4E. the design of experiments is very persuading, especially the 4EBP1 point mutant and pull down of eIF4E give a strong proof to unravel the crucial role of Thr46 phosphorylation in binding eIF4E. Thank the authors for providing the powerful evidences to make me under the mechanism of the interaction between 4EBP1 and eIF4E.
The eIF4E are released from the 4EBP1 in many malignant tumor due to the hyperphosphorylation of 4EBP1. It will be very helpful to discover some new mTOR signal inhibitors and to explain the mechanism, if the processes of 4EBP1 dephosphorylation are clarified.
The paper clearly elucidate the functionally important role of the phosphorylated Thr46 of 4EBP1 in the course of 4EBP1 binding with eIF4E. the design of experiments is very persuading, especially the 4EBP1 point mutant and pull down of eIF4E give a strong proof to unravel the crucial role of Thr46 phosphorylation in binding eIF4E. Thank the authors for providing the powerful evidences to make me under the mechanism of the interaction between 4EBP1 and eIF4E.
The eIF4E are released from the 4EBP1 in many malignant tumor due to the hyperphosphorylation of 4EBP1. It will be very helpful to discover some new mTOR signal inhibitors and to explain the mechanism, if the processes of 4EBP1 dephosphorylation are clarified.
Competing Interests:No competing interests were disclosed.Close
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
The eIF4E are released from the 4EBP1 in many malignant tumor due to the hyperphosphorylation of 4EBP1. It will be very helpful to discover some new mTOR signal inhibitors and to explain the mechanism, if the processes of 4EBP1 dephosphorylation are clarified.
The eIF4E are released from the 4EBP1 in many malignant tumor due to the hyperphosphorylation of 4EBP1. It will be very helpful to discover some new mTOR signal inhibitors and to explain the mechanism, if the processes of 4EBP1 dephosphorylation are clarified.