Keywords
air pollution
air pollution
One common neurodegenerative disease, Parkinson’s disease, has been linked to exposure to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) and to inhaled manganese1,2. Similarly, inhaled aluminum dust has been associated with neurotoxic effects and pre-clinical cognitive impairment3. Certain inhalation anesthetics have also been implicated in elevating AD risk, possibly by exacerbating the neurotoxic oligomerization of the amyloid-β (Aβ) peptide4. The early involvement of the olfactory cortex in AD has caused longtime speculation that some inhaled agent might play a role in AD risk5.
Recently, AD pathology was identified in young people living in areas with high levels of air pollution6,7. Furthermore, impaired cognition has been recently attributed to air pollution exposure in some populations8. These converging lines of evidence led us to analyze brain levels of Aβ40 and Aβ42 in mice exposed to an inhaled particulate matter (nickel nanoparticle; Ni NP) model of air pollution.
All procedures involving animals were conducted in compliance with guidelines for ethical animal research and approved by the New York University School of Medicine Animal Care and Use Committee. Two-month-old male and female FVBN mice (Taconic Farm, Hudson, NY) were randomly assigned to Ni NP inhalation (count median diameter 54 nm, at 1 mg/m3, which is the current Occupational Safety and Health Administration’s Permissible Exposure Limit for nickel hydroxide [http://www.osha.gov/pls/oshaweb/owadisp.show_document?p_table=standards&p_id=9992]) (n = 16 per group) or control filtered air (n = 5 per group) for 3 hours in a nose-only exposure chamber. This protocol has been established as a model for air pollution toxicity in pulmonary disease9, atherosclerosis10, and insulin resistance11. Twenty-four hours post exposure, mice were given pentobarbital, bled out via the vena cava, and then their brains were harvested, snap frozen and stored at -80ºC until assay. For measurement of endogenous mouse brain Aβ40 and Aβ42, we employed the Schmidt method12 and human/rat Aβ 1–40/1–42 ELISA kits (Wako, Richmond, VA). Statistical analysis was performed via Mann-Whitney test. #8 Ni NP is excluded from the analysis due to being more than 2 SD's away from mean or closest value.
Both endogenous Aβ40 and Aβ42 were elevated in the brains of mice following Ni NP exposure (Figure 1). Aβ40 was increased by 1.72-fold (P = 0.0011, Mann-Whitney test), and Aβ42 was increased by 2.29-fold (P = 0.0005, Mann-Whitney test). Aβ42/40 ratio was also increased in the Ni NP-exposed group compared to the filtered air control group (0.27 ± 0.01 and 0.21 ± 0.007, respectively; P = 0.0093, Mann-Whitney test). Both male and female mice responded similarly to Ni NP exposure (male vs. female for Aβ40 and Aβ42 levels; P > 0.1, Mann-Whitney test).
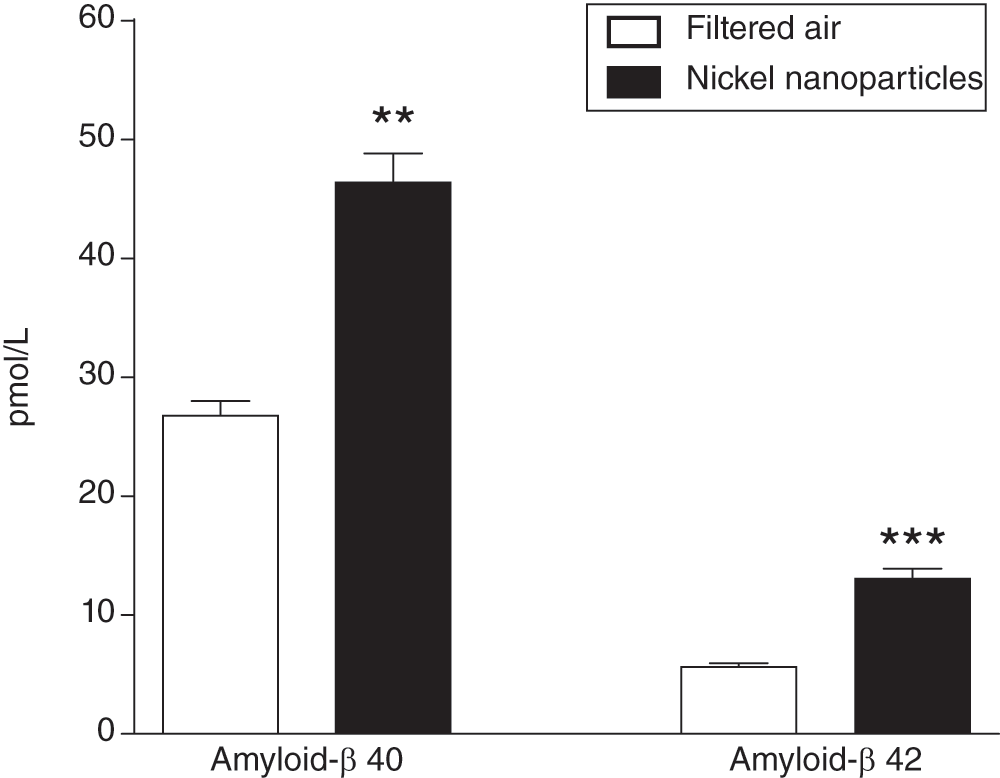
Elevated endogenous mouse brain Aβ40 and Aβ42 in mice exposed to nickel nanoparticles (count median diameter 54 nm, at 1 mg/m3) (n = 16 per group) versus filtered air (n = 5 per group) for 3 hours in a nose-only exposure chamber. Data presented as mean + SEM. **P < 0.01, ***P < 0.001 (Mann-Whitney test).
These data add credence to the proposal4 that one or more inhaled neurotoxin(s) might increase the risk for AD by elevating levels of brain Aβ. We have not identified whether this accumulation occurs at the level(s) of transcription, translation, or post-translational processing. It is tempting to speculate that the well-known links between inhaled toxins and brain inflammation, and other links between brain inflammation and AD established by Griffin and colleagues13 may underlie these phenomena.
The changes that we observed were dramatic, rapid, and unexpected. Human Aβ is more aggregatable than murine Aβ, making it conceivable that the effect on Aβ levels in human brain could be even greater. While elucidating the genesis and molecular underpinnings will be an important next step, an even more important step will be a rigorous application of environmental toxicology and epidemiology to determine whether the elevated brain Aβ caused in mice by this air pollution model corresponds to any situation of authentic human inhalation exposure that is linked to an increased risk for AD.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)