Introduction
Research on model organisms is crucial for discovering the function of genes and DNA elements and for understanding the phenotypic effects of mutations on these genes, which is leading to a better understanding of the pathobiology of human disease1,2. The amount of phenotypic information derived from targeted mutations and hypothesis-driven studies is increasing rapidly, and is now being further augmented by high-throughput international efforts to systematically analyse the effects of genomic variation on model organism phenotypes. For example, the International Mouse Phenotyping Consortium (IMPC3), is undertaking systematic phenotyping studies of the knockouts generated by the International Knockout Mouse Consortium (IKMC4). This means that there will soon be structured phenotype data for loss-of-function mutants for every protein-coding gene in the mouse. Similar approaches are being taken in zebrafish (Danio rerio) by the Zebrafish Mutation Project (ZMP, http://www.sanger.ac.uk/Projects/D_rerio/zmp/) and the data is being made available through the The Zebrafish Model Organism Database (ZFIN5).
Model organism phenotype/genotype datasets are extremely valuable as they can provide clues to human gene functions and involvement in disease processes where no data is available for the human ortholog. At the time of writing, 2,358 human genes are associated with Mendelian phenotypes, but more importantly there are 5,492 human genes with no such phenotype associations, where an orthologous mouse or zebrafish gene does have phenotype data (Data obtained by analysing the file HSgenes_crossSpeciesPhenoAnnotation.txt from http://purl.obolibrary.org/obo/hp/uberpheno/). We have previously demonstrated the power of this approach in determining likely pathogenicity of genes within the intervals of recurrent copy number variation (CNV) diseases6 and it can be applied much more widely in, for example, prioritizing candidate genes identified through human genome wide association studies (GWAS)7,8. Historically, a major problem has been the lack of common semantics across databases, with each project using some combination of free-text descriptions or in-house vocabularies. Thus, phenotype information is not easily integrated across different species. This inhibits comparisons based on phenotype alone, and where orthology is useful phenotypic comparisons cannot be used to their full potential. This is made even more complicated by different conceptualizations of phenotypes in different species and the impact of species-specific anatomies. As the ability of investigators to mobilise this growing collection of model organism data has become more important, it is crucial to develop appropriate ontologies and computational strategies to describe phenotypes such that phenotype descriptions can be objectively related to each other, both within and between species. This becomes even more important as the divergence between the number of human genes with phenotype information and the amount of systematically phenotyped model organism genes is expected to increase in the near future due to high throughput-screens1.
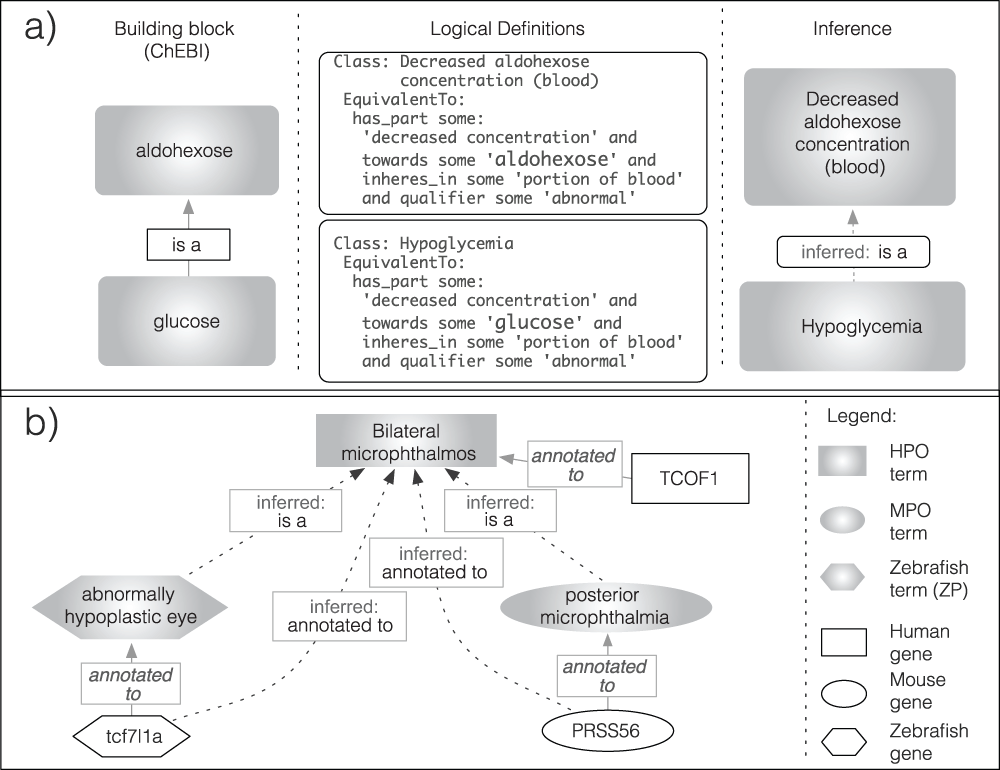
The application of controlled vocabularies and ontologies has accelerated over recent years; the Gene Ontology (GO9) being probably the most successful example in the field of biomedical ontologies. Many other ontologies exist, each of which has been developed for a specific domain in biomedicine. Now a major goal is to increase semantic and syntactic interoperability between those ontologies (e.g. the Open Biomedical Ontologies (OBO) Foundry10). One approach is to develop ontologies by defining complex ("pre-composed") classes in terms of other more elementary (atomic) classes (building blocks) that are species-agnostic. If several ontologies make use of shared building block ontologies, interoperability can be facilitated across a larger domain. For example ontologies that contain classes concerned with DNA-replication in different organisms or cells should refer to a shared class representing DNA-replication-process, enabling computers to detect that the same class is referenced.
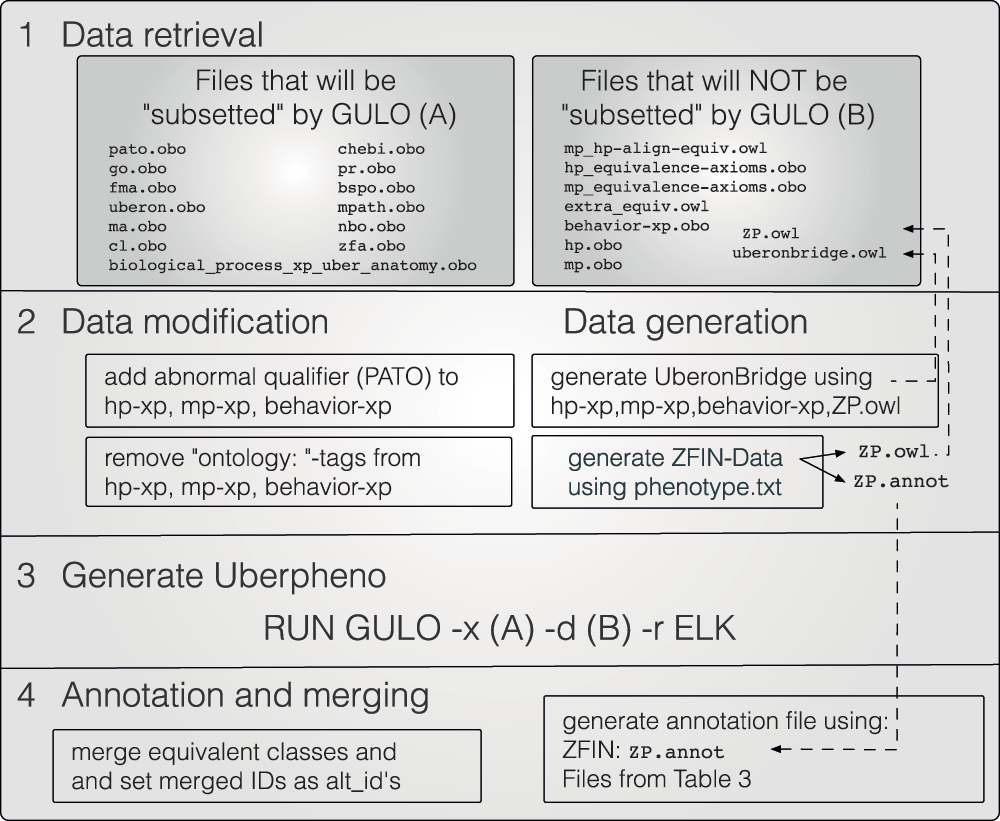
We have previously shown how phenotype information can be linked and used in cross-species phenotype analyses11–15. A crucial part of this strategy is the use of logical definitions to render ontology terms in a way that is computable. Recently, logical definitions of terms representing classes of phenotypic deviations have been developed by several groups. Developers of OBO Foundry ontologies, such as the GO16, the Mammalian Phenotype Ontology (MPO17), the Human Phenotype Ontology (HPO18,19), the Worm Phenotype Ontology20, and also the Cell Ontology21, are now creating logical definitions of their ontology-classes using terms from other building block ontologies. In this effort the Phenotype, Attribute and Trait Ontology (PATO), an ontology of phenotypic qualities, is a key tool19,22. Examples for building block ontologies that are used for the representation of classes of phenotypic abnormalities are given in the upper part of Table 1.
Table 1. Typical building block ontologies: here the focus lies on ontologies that can be used to represent complex classes of phenotype abnormalities in zebrafish, mouse, and human.
| Domain | Name (Abbreviation, Reference) | Downloaded file (relative to http://purl.obolibrary.org/obo/) |
|---|---|---|
| biochemistry | Chemical Entities of Biological Interest (ChEBI29) | chebi.obo |
| Gene Ontology (GO30) | go.obo | |
| proteins | Protein Ontology (PRO31) | pr.obo |
| cell types | Cell Ontology (CL32) | cl.obo |
| anatomy | Foundational Model of Anatomy (FMA33) | fma.obo |
| Spatial Ontology (BSPO-) | bspo.obo | |
| Mouse adult gross anatomy (MA34) | ma.obo | |
| Zebrafish anatomy and development (ZFA35) | zfa.ob | |
| Multi-species anatomy (UBERON36) | uberon.obo | |
| phenotype | Phenotype, Attribute and Trait Ontology (PATO22) | pato.obo |
| Mouse Pathology (MPATH37) | mpath.obo | |
| Mammalian Phenotype Ontology (MPO17) | mp.obo | |
| Human Phenotype Ontology (HPO18) | hp.obo | |
| Neuro Behavior Ontology (NBO38) | nbo.obo |
Comments on this article Comments (0)