Keywords
protein, docking ligand, congruence
protein, docking ligand, congruence
The ability to computationally predict protein-ligand interactions with accuracy is an invaluable asset, since it allows for large scale screening at minimal costs1,2. Consequently, computational methods that predict the favorable conformation of a protein-ligand complex have been the focus of intense research over the last few decades3. Protein docking methods are a subset of these methods, characterized by their ability to score a large number of possible conformations using fast algorithms. Among these, five programs - AutoDock4, GOLD5, DOCK6, FlexX7 and Glide8 - are the most cited, although it should be noted that ‘the number of citations of a given paper is no measure of quality of the corresponding protein-ligand docking software program’9. Typically, a protein-ligand docking program has two distinct phases - conformational sampling (or searching) and scoring10. Despite the significant progress in the field, there are several challenges arising from protein or ligand flexibility, entropic considerations or the presence of water molecules that need to be addressed11.
Previously, the conservation of spatial and electrostatic properties in cognate pairs of residues in the catalytic site of proteins with the same functionality has been used to develop a computational method (CLASP) for detecting binding and catalytic sites12–15. In the current work, this methodology has been extended by proposing a method for docking ligands into target proteins - DOCLASP (Docking using CLASP). DOCLASP takes as input a set of proteins with known structures which bind a particular ligand, and a target protein into which the ligand is to be docked. Each of these holo structures is used to define a motif consisting of the first four residues making non-hydrophobic interactions. These motifs are used to query the target protein, using an enhanced version of the search engine used by CLASP that uses precompiled databases16, and significant congruent matches are identified. These significant matches in the target protein are now superimposed to the binding residues (the motif) in the corresponding holoenzyme(s), thus creating a unified coordinate framework formed by the holoenzyme, the ligand and the target enzyme. This gives us the docked ligand to the target protein, which is outputted as a Pymol formatted file. In essence, the holoenzyme is replaced with the target enzyme if the contact points have a good spatial and electrostatic match in the target enzyme by aligning the congruent atoms. Thus, DOCLASP leverages the implicit search and scoring functions in CLASP to rank possible conformations.
The native activity of phosphoinositide-specific phospholipase C (PI-PLC) was previously shown to be inhibited by two dipeptidyl peptidase-IV (DPP4) inhibitors - vildagliptin (LAF-237) at micromolar concentrations, and K-579 at nanomolar concentrations using in vitro experiments based on CLASP analysis15. Since ‘comparing docking programs can be difficult’9, the DOCLASP methodology is validated by docking vildagliptin to the PI-PLC structure in complex with myo-inositol17. The docked ligand is free from steric clashes and interacts with the exact side chain residues that bind myo-inositol, providing corroboration of the validity of the proposed methodology. Thus, the current work presents a fast methodology for docking ligands into protein structures based on spatial and electrostatic congruence of known binding sites to putative binding targets.
DOCLASP uses the basic hypothesis of CLASP - the non-triviality of the spatial and electrostatic congruence in cognate pairs seen across different structures of the same catalytic function, which is extended to the related concept of ligand binding12. It takes as input a set of M proteins with known structures (Equation 1) which bind a particular ligand (Lig), and a target protein into which Lig is to be docked (Ptarget). Each of these M holo structures is used to define a motif consisting of N (=4) residues (Equation 2), taking the first four closest non-hydrophobic interactions into account (Algorithm 1).
Input: Protein :
Input: Ligand :
Input: n: number of closest atoms to choose
Output: ϕmotif = {atom1 … atomn}
begin
/* Output Motif */
ϕmotif = ∅ ;
/* Accepted atom pairs - exclude hydrophobic interactions*/
ϕAcceptedAtomPair = [O-N, N-O, O-H, H-O, O-O, N-N, N-H, H-N, S-H, H-S] ;
ATOMSLig = atoms of all residues of Ligand ;
/* Initial radius in Å */
Radius = 2.5 ;
foreach atomi in ATOMSLig do
ϕatoms = ProteinAtomsWithinRadiusOfLigandAtom(Protein,atomi, Radius);
foreach atomj in ϕatoms do
if atomi-atomj is in ϕAcceptedAtomPair then
InsertInMotifSet(atomj, ϕmotif);
if (ϕmotif == n) then
last ;
end
end
end
/* increment radius by 0.1 Å */
Radius = Radius + 0.1;
end
return ϕmotif ;
end
Each position of the motif has a set of amino acids specified to allow for stereochemically equivalent matches at that particular position (Equation 3), such that while matching amino acid type of ri should belong to GROUPi.
Previously, the K sets of N residues were obtained in Ptarget using an exhaustive search procedure similar to the one used in SPASM18. An enhanced algorithm now precompiles all possible motifs of a set (N=4 in this case) of predefined amino acid residues from a protein structure that occur within a specified distance16, and selects the appropriate ones based on each motif (Equation 4). Any match below a user defined threshold score (Sthresh) is discarded.
In Case is null, the ligand Lig can not be docked to the target protein Ptarget. , the first element, has the minimum CScore and represents the putative binding site in Ptarget based on the holoenzyme Pi. The set of putative binding sites Φbindsite is thus defined (Equation 5).
Each element of Φbindsite () is now superimposed to the corresponding holoenzyme, based on the motif binding Lig in Pi. In order to superimpose these motifs, linear and rotational transformations are applied on all atoms such that the first three atoms lie on the same plane (Z=0), the first atoms are the origin of the coordinate axis and the second atoms lie on the Y axis. This creates a unified coordinate framework having the holoenzyme, the ligand and the target enzyme, thus providing the docked ligand in the target enzyme. Essentially, the holoenzyme is replaced with the target enzyme if the contact points have a good spatial and electrostatic match in the target enzyme by aligning the congruent atoms. This docked ligand is now outputted as a Pymol formatted file.
The DOCLASP package is written in Perl on Ubuntu. Hardware requirements are modest - all results here are from a simple workstation (2GB ram) and runtimes were a few minutes at the most. Adaptive Poisson-Boltzmann Solver (APBS) and PDB2PQR packages were used to calculate the potential difference between the reactive atoms of the corresponding proteins19,20. The APBS parameters and electrostatic potential units were set as described previously in12. All protein structures were rendered by PyMol (http://www.pymol.org/).
Previous CLASP analysis of the spatial and electrostatic properties of active site residues in PI-PLC from B. cereus indicated that it is a prolyl peptidase, which was also validated by in vitro experiments13. Subsequently, it was shown that PI-PLC is inhibited by two dipeptidyl peptidase-IV (DPP4) inhibitors - vildagliptin (LAF-237) at micromolar concentrations, and K-579 at nanomolar concentrations. Since there are no DPP4 structures solved which ligand K-579, a DPP4 protein structure in complex with vildagliptin (PDBid:3W2TA)21 provided the five closest atoms in the protein (E205, E206, S630, Y662 and Y547) (see Methods) that make non-hydrophobic interactions with the ligand (Table 1).
Interactions are sorted based on the distance. R/A/LA/D: Residue number/Atom of the residue/Atom of ligand/distance between the interacting atoms (in Å). For example, ‘S630/OG/N2/2.4’ means that the atom OG from Ser630 is at 2.4 Å from the N2 atom of vildagliptin in PDBid:3W2TA.
| R/A/LA/D | R/A/LA/D | R/A/LA/D | R/A/LA/D | R/A/LA/D |
|---|---|---|---|---|
| S630/OG/N2/2.4 | E205/OE1/N12/2.8 | Y662/OH/O20/3 | E206/OE2/N12/3 | Y547/OH/N2/3.1 |
A subset of these atoms might be sufficient to ligand vildagliptin in the target protein. Thus, motifs, each with four atoms, were created using the five closest atoms in the protein. The binding of ligands is known to induce electrostatic and spatial perturbations in the binding site. The spatial and electrostatic perturbations induced by the vildagliptin binding is shown by comparing the apo (PDBid:2OQIA) and the holoenzyme (PDBid:3W2TA) in Table 2. Hence, the electrostatic and spatial profile of the motif were obtained from the apo DPP4 enzyme (PDBid:2OQIA), and then used for querying the PI-PLC apo structure (PDBid:1PTDA).
We compare the pairwise distance and electrostatic potential difference (EPD) changes in the apo (PDBid:2OQIA) and holo (PDBid:3W2TA) enzymes. Note that the pairwise distance between these atoms change in the ligand free PDB (2OQIA) as compared to the protein with bound inhibitor (2BUBA). For example, the distance between E205OE2 and E206OE2 (pair ab) changes from 3.9 Å to 5.6 Å. Also, there is a definite change in the EPD between E205OE2 and E205OE2 (pair ab). D = Pairwise distance in Å. PD = Pairwise potential difference. The electrostatic potential are in dimensionless units of kT/e where k is Boltzmann’s constant, T is the temperature in K and e is the charge of an electron.
These motifs were used to query the PI-PLC structure using an enhanced algorithm (PREMONITION) that precompiles all motifs in a database16. Table 3 shows the best matches obtained in the PI-PLC structure for the five partial motifs. All these matches have significant electrostatic congruence. The root mean square deviation (RMSD) have low values - however, this is a deceptive metric since these deviations are averaged out. The maximum pairwise distance is another metric to discriminate the spatial congruence, and should be used in combination with the RMSD value. All of these above mentioned matches have significant maximum pairwise distance deviation. Also, three matches do not comprise of active site residues (motifs 2, 3 and 4). However, it can be seen that the first and fifth matches comprises of active site residues (involved in the binding of myo-inositol in PDBid:1PTGA), and have three residues (Asp67, Asp198 and Trp178) in common.
The comparison is done using apo enzymes (PDBid:1PTDA for PI-PLC and PDBid:2OQIA for DPP4), since the binding of a ligand induces spatial and electrostatic changes in the active site. The fifth motif has the least rmsd deviation, and comprises of active site residues (involved in the binding of myo-inositol in PDBid:1PTGA). Out of the best matches in the other four motifs, three do not comprise of active site residues (motifs 2, 3 and 4). Motif 1 has a reasonably significant match, and has three residues (Asp67, Asp198 and Trp178) in common with the best match for Motif 5. These three residues from PI-PLC (Asp67, Asp198 and Trp178) and corresponding three residues from DPP4 (Glu205, Glu206 and Tyr662) were used to superimpose DPP4 and PI-PLC. N = Motif number. D = Pairwise distance in Å. PD = Pairwise potential difference. Rmsd = Root mean square deviation. Max = maximum pairwise distance deviation. APBS writes out the electrostatic potential in dimensionless units of kT/e where k is Boltzmann’s constant, T is the temperature in K and e is the charge of an electron.
Thus, these three residues from PI-PLC (Asp67, Asp198 and Trp178) and corresponding three residues from DPP4 (Glu205, Glu206 and Tyr662) were used to superimpose DPP4 and PI-PLC. Table 4 shows the congruence of these residues. The corresponding holo structures - PDBid:3W2TA for DPP4, and PDBid:1PTGA for PIPLC - were used for the superimposition. This superimposition applies geometric transformations such that Asp67OD1 and Glu205OE2 were at the center of the coordinate axis (coordinates = [0,0,0]), Asp198OD1 and Glu206OE2 lies on the X-Y axis (i.e. Y coordinate is 0) and Tyr662CZ and Trp178CZ2 were on the X-Y plane (i.e. Z coordinate is 0). Figure 1 shows the superimposed proteins. It is observed that (Asp67, Asp198 and Trp178) overlaps well with (Glu205, Glu206 and Tyr662).
The match is significant and comprises active site residues (involved in the binding of myo-inositol in PDBid:1PTGA). Pair ‘ab’ is considered to be electrostatically congruent since the PD values are close to zero, and can be considered almost equipotential. This is expected for atoms of the same type from the same residue (GLU205OE1/GLU206OE1 and ASP67OD1/ASP198OD1). These three residues from PI-PLC (Asp67, Asp198 and Trp178) and corresponding three residues from DPP4 (Glu205, Glu206 and Tyr662) were used to superimpose DPP4 and PI-PLC. D = Pairwise distance in Å. PD = Pairwise potential difference. APBS writes out the electrostatic potential in dimensionless units of kT/e where k is Boltzmann’s constant, T is the temperature in K and e is the charge of an electron.
| PDB | Atoms(a,b,c) | ab | ac | bc | |
|---|---|---|---|---|---|
| 2OQIA 1PTDA | GLU205OE1,GLU206OE1,TYR662CZ, ASP67OD1,ASP198OD1,TRP178CZ2, | D PD D PD | 6.6 -9.3 7.7 81.8 | 7.2 -341.5 6.7 -275.7 | 5.8 -332.2 4.9 -357.6 |
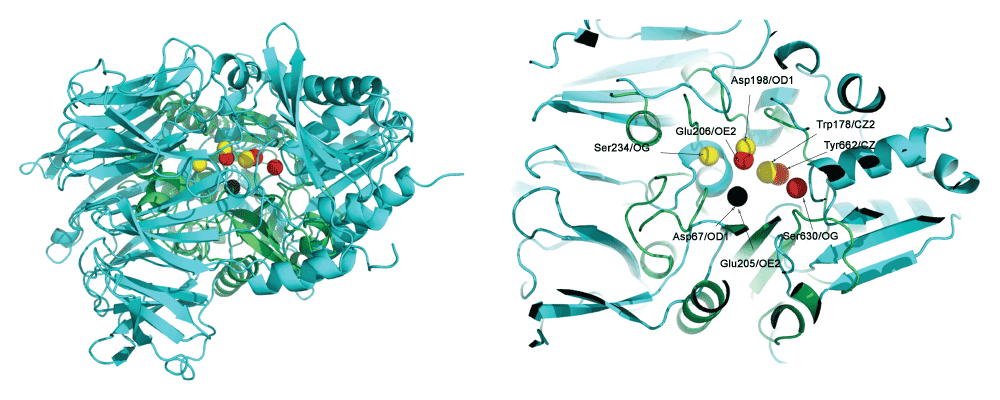
Three residues from PI-PLC (Asp67, Asp198 and Trp178 in yellow) were superimposed to the corresponding three residues from DPP4 (Glu205, Glu206 and Tyr662 in red). Asp67OD1 and Glu205OE2 is at the center of the coordinate axis (coordinates = [0,0,0]) (in black), Asp198OD1 and Glu206OE2 lies on the X-Y axis (i.e. Y coordinate is 0) and Tyr662CZ and Trp178CZ2 are on the X-Y plane (i.e. Z coordinate is 0). Asp67, Asp198 and Trp178 in the PI-PLC protein overlaps well with Glu205, Glu206 and Tyr662 from DPP4, but the Ser234-Ser630 pair is not spatially congruent.
These transformations were also applied to the vildagliptin molecule, and this resulted in a docked structure for this molecule into the PI-PLC protein. Figure 2 shows the vildagliptin docked into the PI-PLC structure which is complexed with myo-inositol (PDBid:1PTGA). The distances of the atoms in vildagliptin and myo-inositol that interact (excluding hydrophobic interactions) to the first ten residues in the PI-PLC structure are shown in Table 5. It is interesting to note that the residues shown in Table 5 are all part of side chain residues in close contact with the myo-inositol ring (shown in Figure 713). Further validation was obtained by observing that both Arg69/NH1 and His32/NE2 interact with atom O4 in vildagliptin, and Arg69/NH2 and His32/NE2 interact with O2 in myo-inositol in the PI-PLC structure. The Pymol script for visualizing the docking (SupplementaryPymol.p1m) and a movie (SupplementaryMovie.avi) are also provided as Supplementary information.
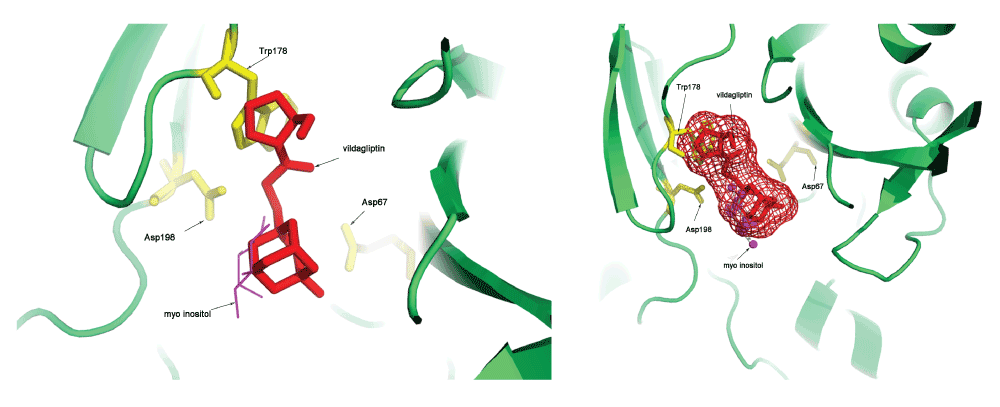
It can be seen that vildagliptin fits into the binding site of PI-PLC. It also makes non-hydrophobic contacts to the residues in the protein similar to those made by myo-inositol (Table 5).
These interactions exclude hydrophobic interactions, and the first closest ten atoms are chosen. Out of ten, seven residues obtained by docking vildagliptin using DOCLASP are seen to be equivalent to those that are known to bind myo-inositol to the PI-PLC structure13, while two more have the same amino acid type (marked by asterisks). Only one pair has a different amino acid type (Tyr200 for myo-inositol and Glu117 for vildagliptin). Further validation is obtained by observing that both Arg69/NH1 and His32/NE2 both interact with atom O4 in vildagliptin, and Arg69/NH2 and His32/NE2 interact with O2 in myo-inositol in the PI-PLC structure.
It is important to comment on the previous hypothesis of a nucleophilic serine being responsible for the inhibition of PI-PLC using vildagliptin. The electrostatic and spatial profile of the motif 4 from DPP4 is compared to the electrostatic and spatial profile of matching active site residues in PI-PLC in Table 6, including serine in the comparison. It can be seen that Ser630 in DPP4 has a significant spatial difference as compared to Ser234 in PI-PLC (pair ‘bc’ has a difference of 5 Å), and also a reasonable electrostatic difference (pair ‘ac’ has a difference of 144 PD units). The relatively large distance over which Ser234 in PI-PLC interacts with myo-inositol (4.8 Å) indicates that Ser234 is not directly involved in the binding of the ligand. However, it is responsible for creating the electrostatic milieu that is required for other interacting residues to attain their appropriate potential. Even for DPP4, many inhibitors do not interact with the nucleophilic Ser63022 - although the vildagliptin molecule does21. Thus, the previous conjecture of a nucleophilic serine being directly responsible for the binding of DPP4 inhibitors to PI-PLC, as implied by the catalytic triad congruence, is incorrect13. However, this serine is indirectly responsible for driving the neighboring residues to an appropriate state. Spatial constraints are an additional discriminator.
Both the structures are apo enzymes, since the binding of a ligand induces spatial and electrostatic changes in the active site. Ser630 in DPP4 has a significant spatial difference as compared to Ser234 in PI-PLC (pair ‘bc’ has a difference of 5 Å), and also a reasonable electrostatic difference (pair ‘ac’ has a difference of 144 PD units). D = Pairwise distance in Å. PD = Pairwise potential difference. APBS writes out the electrostatic potential in dimensionless units of kT/e where k is Boltzmann’s constant, T is the temperature in K and e is the charge of an electron.
The number of docking methods available is such that even a detailed review could only provide a partial list of currently available docking methods9. The current work presents a template based, static method that leverages the spatial and electrostatic properties of the binding site. The definitive advantage of a static method can only be highlighted by emphasizing the known limitations of conformational sampling of the protein structure23,24. The problem is indeed exacerbated by the plasticity of the drug itself25–27. While DOCLASP is completely ineffectual in the absence of such a database, unlike de novo methods, it benefits from the burgeoning database of protein-ligand structures28. The conservation of electrostatic properties, extracted using APBS/PDB2PQR, is the strongest argument in favor of DOCLASP.
There are several limitations in the method. Firstly, it can be applied to those compounds which are bound to proteins whose structures have been solved. Additionally, it is requires the structure of the apoenzyme, as this is used to extract the query motif considering the structural and electrostatic changes induced by ligand binding. However, with an ever increasing number of protein structures being solved, this is not a severe limitation since most proteins with ligands also have their apo structures solved. Furthermore, the lack of congruent matches leads DOCLASP to return a null result. This can be overcome by relaxing constraints, for example by checking for spatial congruence only. Finally, it is required to develop an energy function which will be able to discriminate poorly docked structures that have either significant steric clashes or are docked on the surface of the protein.
To summarize, this work presents an implicit method for docking ligands to proteins, in which the search and scoring are implicit in the CLASP algorithm. One significant limitation of this method is the requirement of template protein structures in complex with the given compound. As future work, I intend to incorporate the flexibility of the ligand and protein to add further discrimination.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)