Leanza V, Rubbino G and Leanza G. Atypical Cornelia de Lange Syndrome: a case report [version 1; peer review: 2 approved with reservations]. F1000Research 2014, 3:33 (https://doi.org/10.12688/f1000research.3-33.v1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
1Surgery Department, Obstetrics and Gynecologic Unit “Ospedale Santo Bambino”, Catania University, Catania, Italy
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
Cornelia de Lange Syndrome (CdLS) (also called Bushy Syndrome or Amsterdam dwarfism), is a genetic disorder that can lead to several alterations. This disease affects both physical and psychical development. The various abnormalities include facial dysmorphia (arched eyebrows, synophrys, depressed nasal bridge, long philtrum, down-turned angles of the mouth), upper-extremity malformations, hirsutism, cardiac defects, and gastrointestinal alterations. The prevalence of this syndrome is approximately one per 15,000. Ultrasound is not the perfect means to diagnose CdLS, however, many abnormalities can be detected prenatally by scrupulous image observation. We report an atypical CdLS case characterized by increased nuchal translucency in the first trimester, normal kariotype, saddle nose, micrognathia with receding jaw, low set ears, facies senilis, arthrogryposis of the hands, absence of the Aranzio ductus venous, dilatation of gallbladder and bowel, a unique umbilical artery, increased volume of amniotic fluid, and intrauterine growth retardation ending with the interruption of pregnancy.
Cornelia de Lange Syndrome (CdLS) is a genetic disorder that can lead to several alterations. It affects both physical and psychical development. The several abnormalities include facial dysmorphia (arched eyebrows, synophrys, depressed nasal bridge, long philtrum, down-turned angles of the mouth), upper-extremity malformations, hirsutism, cardiac defects, and gastrointestinal alterations1. The prevalence of this syndrome is approximately one per 15,0002. Many markers have to be considered. Nuchal translucency (NT) measurement during the first trimester screening between 11 and 14 weeks gestation has now been clearly identified as a marker for aneuploidies and in particular for trisomy 21. Even in the absence of aneuploidy, increased fetal nuchal translucency has been shown to be a marker for fetal heart malformations and several other fetal defects linked to genetic syndromes when the measure exceeds the 95th percentile (3–5 mm)3. When conventional karyotyping is normal, enlarged NT is a strong marker for adverse pregnancy outcome, associated with miscarriage or intrauterine death). Unspecified genetic syndromes involving developmental delay may only emerge after birth and become evident after the first years of life. Several abnormalities have been reported in fetuses with enlarged NT: the majority are cardiac defects, diaphragmatic hernia, exomphalos, body stalk anomaly, skeletal defects, certain genetic syndromes (such as congenital adrenal hyperplasia), fetal akinesia, deformation sequence, Noonan syndrome, Smith-lemli-Opitz syndrome and spinal muscular atrophy4. The saddle nose, characterized by a markedly depressed bridge has been described in AIDS embryopathy, Christ-Siemans-Touraine syndrome, various deletion syndromes, fetal trimethadione syndrome, Laron-type dwarfism, leprechaunism, multiple epiphyseal dysplasia, otospondylomegaepiphyseal dysplasia (OSMED) syndrome, relapsing polychondritis, thanatophoric dwarfism, Wegener’s granulomatosis, and various conditions that are further characterized by gargoyle-like facies. Micrognathia (mandible of insufficient size) is a malformation of the fetal face characterized by a small mandible. Micrognathia may be idiopathic but is more commonly associated with many different syndromes. Retrognathia (recession of the chin) is assessed through the measurement of the inferior facial angle, as defined on a mid-sagittal view. With routine ultrasound, the receded chin may be seen on a profile of the face. Yet, this diagnosis is often missed during a routine ultrasound examination. The mandible is known to grow significantly during the third trimester. If mandibular alteration is suspected, particular attention should be paid to the growth of the chin throughout the remainder of the pregnancy. Conditions associated with micrognathia include chromosomal abnormalities, neuromuscular abnormalities, single-gene disorders, and other syndromes. The prognosis of fetal micrognathia is poor, even in chromosomally normal fetuses. Frequent malformations associated with micrognathia are: Pierre Robin sequence (micrognathia, cleft palate, or both)5; Cerebrocostomandibular syndrome (diagnosed on the basis of micrognathia, a posterior cleft palate defect, and characteristic rib gap abnormalities); Cornelia de Lange syndrome, (underdeveloped chin with tetralogy of Fallot, intrauterine growth restriction, and an abnormal right hand)6; hypochondrogenesis type II and Caffey disease.
Case report
We report a case of a healthy 31-year-old, gravida 2, para 1 at 30 weeks of gestation that was admitted to S. Bambino University Hospital in Catania for ultrasound examination.
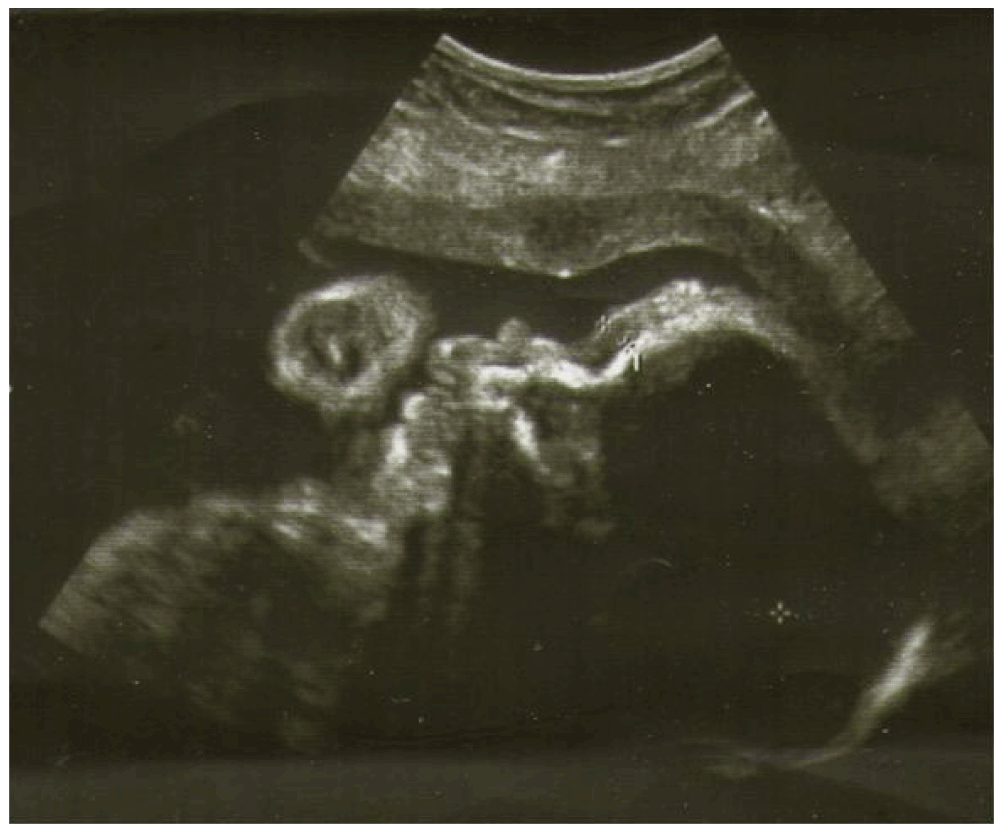
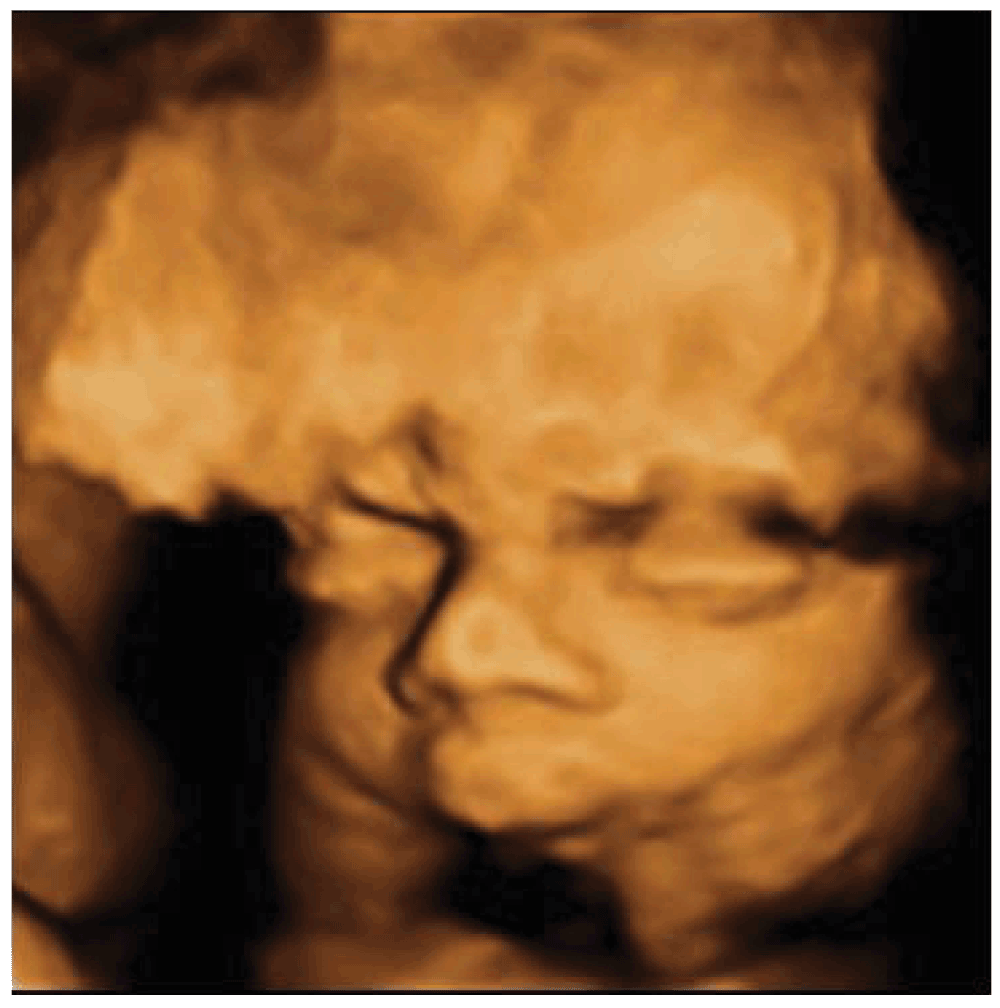
Ultrasounds revealed nuchal edema, saddle nose, micrognathia with receding jaw, low set ears, facies senilis at 3D ultrasounds, arthrogryposis of the hands, absence of Aranzio ductus venous, dilation of gallbladder and bowel, single one umbilical artery, increased volume of amniotic fluid. Intrauterine growth retardation was associated as well. (Figure 1) Micrognathia was evident on midsagittal and 3D scan. The biparietal diameter was 68 mm, femur length 47 mm, suggesting fetal growth restriction. Pulsed Doppler sonography showed normal middle cerebral artery and umbilical artery pulsatility indices. The obstetric history revealed increased nuchal translucency thickness (NT) at 11 weeks (4 mm, > 95 centile). No genetic alterations were found at the amniocentesis carried out during 16th week of pregnancy (normal karyotype of 46, XX). Morphological ultrasound at 22 weeks of pregnancy was not able to detect the syndrome. A further ultrasound examination at 29 weeks (Figure 2) of pregnancy pointed out a fetal dimorphism and the pregnant woman asked for our consultation.
Figure 1. 30th week face profile with ultrasound.
Figure 2. 29thweek face aspect with 3D ultrasound.
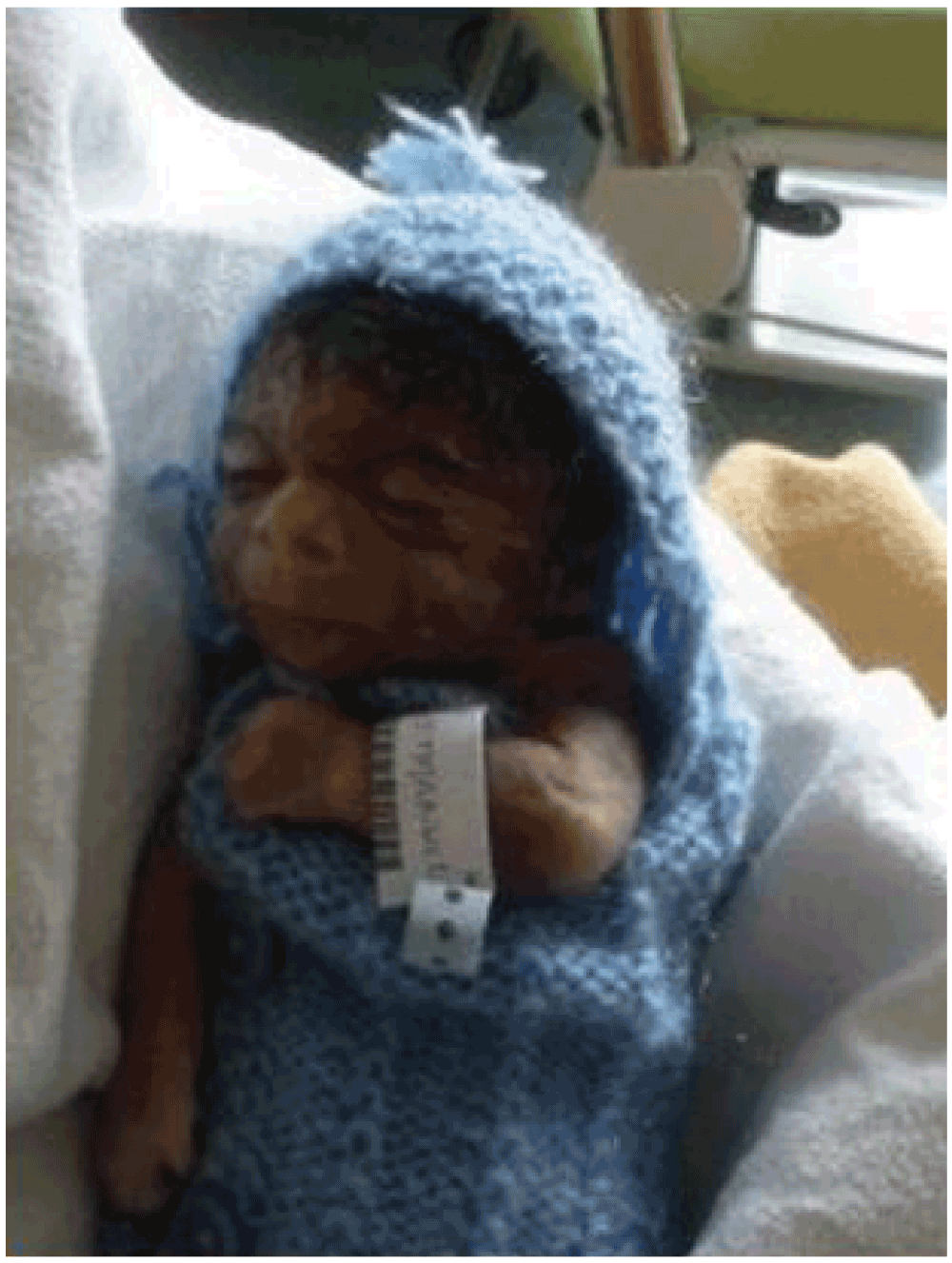
Soon after, interruption of pregnancy occurred. The autopsy showed a foetus with a weight between the 5th and the 10th percentile and a dysmorphic syndrome with malformation features amenable to CdLS (Figure 3). The foetus had a typical dysmorphic face, hirsutism, rhizomelic limb, bilateral camptodactyly, single transverse palmar crease, proximal and very short inches with hypoplasia of 1st metacarpal and single phalanx, II-III membranous syndactyly of feet, right diaphragmatic hernia, bilateral cryptorchidism, microcephaly, numerous nodules of heterotopic cerebellar vermis, single umbilical artery and hypotrophic placenta devoid of inflammatory lesions.
Figure 3. Aspect of the new born (facies senilis).
Discussion and conclusion
CdLS is a multisystem malformation syndrome recognized primarily on the basis of the morphological characteristics (malformations of the cranial, cardiac, gastrointestinal, and skeletal systems)7. However, there is wide clinical variability of disorders, with milder phenotypes that may be difficult to ascertain on the basis of physical features. In certain cases the diagnosis may be missed when ultrasound examination is not performed accurately8. Criteria to detect CdLS are not standardized. The main alterations are as follows9:
Facial abnormalities (synophrys, long eyelashes, microcephaly, anteverted nostrils)
Cardiac defects (defects of ventricles or atria, aortic or pulmonary stenoses, tetralogy of Fallot, atrioventricular canal, single ventricle, aorto-pulmonary window, truncus arteriosus communis)
Abnormalities of upper limbs (ectrodactylia and monodactylism)
Ultrasound detection of eyelashes is considered a clue for prenatal diagnosis of CdLS, but it can be missed in clinical practice10. CdLS is considered a cohesinopathy. Mutations in cohesin, or its regulators, cause a spectrum of human developmental syndromes. Cohesinopathy disorders include both CdLS and Roberts Syndrome. The discovery of novel roles for chromatid cohesion proteins in regulating gene expression led to the idea that cohesinopathies are caused by dysregulation of multiple genes downstream of mutations in cohesion proteins. Consistent with this idea, there is an altered expression of developmental genes and an incomplete overlap among dysregulated genes in different components of the cohesion apparatus11. CdLS is considered a dominantly inherited congenital malformation disorder, caused by mutations in the cohesin-loading protein NIPBL for nearly 60% of individuals. In humans, the multisubunit complex cohesin is made up of SMC1, SMC3, RAD21 and a STAG protein. These form a ring structure that is proposed to encircle sister chromatids to mediate sister chromatid cohesion; it also has a key role in genetic regulation. In CdLS cell lines an altered transcription with either NIPBL or HDAC8 mutations has been found12. The proteins produced from mutated genes interfere in the foetal development. Within cells, these proteins help regulate the structure and organization of chromosomes and are involved in the repair of damaged DNA. They also regulate the activity of certain genes in the developing limbs, face, and other parts of the body. Researchers are looking for additional changes in the NIPBL, SMC1A and SMC3 genes, as well as mutations in other genes, that may be responsible for this condition. The majority of cases result from new gene mutations and occur in people with no history of familiarity. CdLS can be prenatally diagnosed in a family with a known mutation in a CdLS gene. The characteristic ultrasonographic profile may allow for prenatal diagnosis of CdLS in subsequent pregnancies for couples with a prior child with CdLS in whom a mutation has not been identified or when there are unexplained signs of fetal abnormality during pregnancy, such as oligo- or polyhydramnios, a low maternal serum PAPP-A level and/or increased NT, fetal growth retardation, or structural anomalies consistent with CdLS13. In some cases the diagnosis is made prenatally in other cases the syndrome is diagnosed after childbirth.
The diagnosis is seldom in doubt when there is a major longitudinal deficiency defect of the upper limb, severe prenatal and postnatal growth retardation, and severe mental retardation. Features used to make the correct diagnosis include penciled and arched eyebrows, a high set/short anteverted nose, a long flat philtrum, a thin upper lip, downturned corners of the mouth, and micrognathia. Characteristics that are misleading include full or flat brows, a prominent nasal bridge or bulbous tip, and/or a normal or prominent chin. As genetic and biochemical tests are unreliable at present, antenatal detection depends upon identification of some aspects of the phenotype in the fetus using ultrasound imaging14,15. When the syndrome is not recognized during pregnancy, the newborn may survive with a low quality of life and, thus, medical team could become involved in surgical procedures16. Each malformation causes an impact in the psychological sphere of both the individual and the family17,18. Finally, last but not the least, the failure of an early diagnosis may lead to medical-legal issues19.
Consent
Written informed consent for publication of clinical details and clinical images was obtained from the patient.
Author contributions
VL: conception and design. GR: performed the ecography and drafted the article. All authors revised the manuscript for intellectual content and approved the final manuscript for publication.
Competing interests
No competing interests were disclosed.
Grant information
The author(s) declared that no grants were involved in supporting this work.
Acknowledgements
Special thanks to Professor Salvatore Sciarretta for his precious help in translating this work.
Faculty Opinions recommended
References
1.
Noor N, Kazmi Z, Mehnaz A:
Cornelia de Lange syndrome.
J Coll Physicians Surg Pak.
2012; 22(6): 412–3. PubMed Abstract
2.
Murray JE, Walayat M, Gillett P, et al.:
An atypical facial appearance and growth pattern in a child with Cornelia de Lange Syndrome: an intragenic deletion predicting loss of the N-terminal region of NIPBL.
Clin Dysmorphol.
2012; 21(1): 22–3. PubMed Abstract
| Publisher Full Text
3.
Senat MV, Frydman R:
Increased nuchal translucency with normal karyotype.
Gynecol Obstet Fertil.
2007; 35(6): 507–15. PubMed Abstract
| Publisher Full Text
4.
Mula R, Goncé A, Bennásar M, et al.:
Increased nuchal translucency and normal karyotype: perinatal and pediatric outcomes at 2 years of age.
Ultrasound Obstet Gynecol.
2012; 39(1): 34–41. PubMed Abstract
| Publisher Full Text
5.
Hsieh YY, Chang CC, Tsai HD, et al.:
The prenatal diagnosis of Pierre-Robin sequence.
Prenat Diagn.
1999; 19(6): 567–569. PubMed Abstract
| Publisher Full Text
7.
de Lange C:
Sur un type nouveau d degenerescence (typus Amestelodamensis). Archives de Médecine des Enfants. 1933; 36: 713–719.
8.
Bhuiyan ZA, Klein M, Hammond P, et al.:
Genotype-phenotype correlations of 39 patients with Cornelia de Lange syndrome: the Dutch experience.
J Med Genet.
2006; 43(7): 568–575. PubMed Abstract
| Publisher Full Text
| Free Full Text
9.
Akahori Y, Masuyama H, Masumoto Y, et al.:
Three-Dimensional Ultrasound Findings in Cornelia de Lange Syndrome: A Case Report.
Obstet Gynecol.
2012; 2012: 568351. PubMed Abstract
| Publisher Full Text
| Free Full Text
10.
Spaggiari E, Vuillard E, Khung-Savatovsky S, et al.:
Ultrasound Detection of Eyelashes: A Clue for Prenatal Diagnosis of Cornelia de Lange Syndrome.
Ultrasound Obstet Gynecol.
2013; 41(3): 341–2. PubMed Abstract
| Publisher Full Text
11.
Horsfield JA, Print CG, Mönnich M:
Diverse developmental disorders from the one ring: distinct molecular pathways underlie the cohesinopathies.
Front Genet.
2012; 3: 171. PubMed Abstract
| Publisher Full Text
| Free Full Text
12.
Deardorff MA, Bando M, Nakato R, et al.:
HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle.
Nature.
2012; 489(7415): 313–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
13.
Clark DM, Sherer I, Deardorff MA, et al.:
Identification of a prenatal profile of Cornelia de Lange syndrome (CdLS): a review of 53 CdLS pregnancies.
Am J Med Genet A.
2012; 158A(8): 1848–56. PubMed Abstract
| Publisher Full Text
| Free Full Text
14.
Leanza V, Sciarretta S:
Fundamental obstetric and gynecologic elements. (Test Book) Editor S.p.e. Catania, (Italy). 2011; ISBN 978-88-96808-05-4.
16.
Zanghì G, Di Stefano G, Leanza V, et al.:
Incisional hernia in Day Surgery: our personal experience.
G Chir.
2012; 33(6–7): 218–20. PubMed Abstract
17.
Leanza V, Passanisi A, Leanza G:
A specific application of two psychological measures on female urinary incontinence: perceived negative affective self-efficacy scale and stress psychological measure.
Urogynaecologia International Journal.
2011; 25: N°2, 41–48.
18.
Leanza G, Palumbo M, Marilli I, et al.:
Double atresic uterus associated with agenesis of cervix and third upper of vagina. Giornale Italiano di Ostetricia e Ginecologia. 2013; 35(1): 262–264. Reference Source
19.
Coco G, Danzì M, Bellocchi P, et al.:
Medicolegal epidemiological observatory of contentious procedures in the Vittorio Emanuele-Ferrarotto- S Bambino Catania in a University-Hospital (Italy): retrospective analysis of one decade (1995–2005). International Academy of Legal Medicine. 2006; 160–161. Reference Source
Leanza V, Rubbino G and Leanza G. Atypical Cornelia de Lange Syndrome: a case report [version 1; peer review: 2 approved with reservations]. F1000Research 2014, 3:33 (https://doi.org/10.12688/f1000research.3-33.v1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Jackson L. Reviewer Report For: Atypical Cornelia de Lange Syndrome: a case report [version 1; peer review: 2 approved with reservations]. F1000Research 2014, 3:33 (https://doi.org/10.5256/f1000research.1212.r4483)
I agree with the report and comments of Dr Jinglian Liu, who has also reviewed this article. The case report is interesting in its detail of the prenatal ultrasound examination and observations, but completely fails its responsibility to connect those observations
... Continue reading
I agree with the report and comments of Dr Jinglian Liu, who has also reviewed this article. The case report is interesting in its detail of the prenatal ultrasound examination and observations, but completely fails its responsibility to connect those observations to appropriate use of contemporary molecular diagnostic opportunities. As such, this article fails the medical community by not accepting the opportunity to connect their useful prenatal morphological observations to detectable objective molecular findings.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard, however I have significant reservations, as outlined above.
Jackson L. Reviewer Report For: Atypical Cornelia de Lange Syndrome: a case report [version 1; peer review: 2 approved with reservations]. F1000Research 2014, 3:33 (https://doi.org/10.5256/f1000research.1212.r4483)
Liu J. Reviewer Report For: Atypical Cornelia de Lange Syndrome: a case report [version 1; peer review: 2 approved with reservations]. F1000Research 2014, 3:33 (https://doi.org/10.5256/f1000research.1212.r3450)
This manuscript describes a fetus with clinical features suggestive of Cornelia de Lange Syndrome (CdLS).
My comments are as follows:
Data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) would be important for this case. The article
... Continue reading
This manuscript describes a fetus with clinical features suggestive of Cornelia de Lange Syndrome (CdLS).
My comments are as follows:
Data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) would be important for this case. The article published by Clark et al. (2012) which was also cited by the authors, recommends molecular analysis of CdLS genes to prenatal diagnosis of CdLS.
The authors should compare prenatal findings in the current case with what described in the article by Clark et al. (2012). Without molecular testing data, I would like to see a table demonstrating the current case presenting prenatal clinical features consistent with what has been documented in the literature by studying a larger cohort of probands.
In the discussion, in the beginning of first paragraph, the authors claim that CdLS is diagnosed primarily by morphological characteristic and cited a paper published in 1933. At present, the diagnosis criteria of CdLS cannot be the same as what was 90 years ago, particularly with the identification of specific gene mutations in CdLS probands. Please re-phrase this part, and cite updated references.
In the end of the discussion, what does it mean “genetic tests are not reliable”? As a matter of fact, molecular testing has become a widely accepted concept and a popular diagnostic approach for individuals with congenital anormalies.
Minor things:
OMIM numbers of CdLS and disease genes should be given.
Please double check the spelling and eliminate typographic errors, also, some terms are not accurately used. Below is listed some examples of those errors:
Abstract – “kariotype” -> “karyotype”
Introduction – “psychical” - > I’d like to use “neuropsychiatric”
2nd paragraph in case report – “dimorphism” -> “dysmorphism”
2nd paragraph in discussion – “cohesion” -> “cohesin”
2nd paragraph in discussion – please italicize the gene names “NIPBL” and “HDAC8”
2nd paragraph in discussion – “familiarity” ? What does it mean? Why not change to “ no family history”
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard, however I have significant reservations, as outlined above.
Liu J. Reviewer Report For: Atypical Cornelia de Lange Syndrome: a case report [version 1; peer review: 2 approved with reservations]. F1000Research 2014, 3:33 (https://doi.org/10.5256/f1000research.1212.r3450)
vito leanza, Surgery Department, Obstetrics and Gynecologic Unit “Ospedale Santo Bambino”, Catania University, Catania, Italy
01 Sep 2014
Author Response
“Data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) would be important for this case. The article published by Clark et al. (2012) which was
...
Continue reading
“Data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) would be important for this case. The article published by Clark et al. (2012) which was also cited by the authors, recommends molecular analysis of CdLS genes to prenatal diagnosis of CdLS.”
In our case report, data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) are negative , indeed it was called atypical Cornelia de Lange Syndrome (CdLS).
Mutations in NIPBL are not present in all cases and they account for about 60% of CdLS; mutations in SMC1A and SMC3 account for a small percentage (Deardorff et al., 2011).
“The authors should compare prenatal findings in the current case with what described in the article by Clark et al. (2012). Without molecular testing data, I would like to see a table demonstrating the current case presenting prenatal clinical features consistent with what has been documented in the literature by studying a larger cohort of probands.”
Cornelia de Lange Syndrome (CdLS) is a multisystem developmental disorder characterized by growth retardation, cognitive impairment, external and internal structural malformations, and characteristic facial features. Currently, there are no definitive prenatal screening measures that lead to the diagnosis of CdLS. (Clark et al. 2012).
In our case (atypical CdLS) increased nuchal translucency in the first trimester, normal karyotype, saddle nose, micrognathia with receding jaw, low set ears, facies senilis, arthrogryposis of the hands, absence of the Aranzio ductus venous, dilatation of gallbladder and bowel, a unique umbilical artery, increased volume of amniotic fluid, and intrauterine growth retardation were found.
Fewer than 1% of individuals diagnosed with Cornelia de Lange syndrome have an affected parent.
Recommendations for the evaluation of parents of a proband with an apparent de novo mutation include clinical examination for features of CdLS, complete with plotting of growth parameters and molecular genetic testing if the NIPBL mutation has been identified in the proband. In our atypical Cornelia de Lange syndrome molecular tests of parents were negative.
“In the discussion, in the beginning of first paragraph, the authors claim that CdLS is diagnosed primarily by morphological characteristic and cited a paper published in 1933. At present, the diagnosis criteria of CdLS cannot be the same as what was 90 years ago, particularly with the identification of specific gene mutations in CdLS probands. Please re-phrase this part, and cite updated references.”
Diagnosis is based on clinical findings. High-resolution ultrasound examination to follow growth and to evaluate the limbs, heart, diaphragm, palate, and other organs or structures affected in CdLS is fundamental. Reported prenatal ultrasound findings include: increased nuchal translucency in the first trimester [Sekimoto et al., 2000; Huang & Porto, 2002]; Growth failure, which typically presents in the second trimester; and the typical in utero facial profile of a fetus with CdLS consisting of micrognathia, a prominent upper lip, and a depressed nasal bridge with somewhat anteverted nares [Ranzini et al., 1997; Boog et al., 1999; Urban & Hartung, 2001].
“In the end of the discussion, what does it mean “genetic tests are not reliable”? As a matter of fact, molecular testing has become a widely accepted concept and a popular diagnostic approach for individuals with congenital anormalies.”
Genetic tests are not reliable when negative. On the contrary when positive they are important for syndrome.
Indeed, mutations in NIPBL are not present in all cases and they account for about 60% of CdLS; mutations in SMC1A and SMC3 account for a small percentage (Deardorff et al., 2011).
Minor things:
These minor things are easy to correct. I thank the first referee for his clarifications.
“Data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) would be important for this case. The article published by Clark et al. (2012) which was also cited by the authors, recommends molecular analysis of CdLS genes to prenatal diagnosis of CdLS.”
In our case report, data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) are negative , indeed it was called atypical Cornelia de Lange Syndrome (CdLS).
Mutations in NIPBL are not present in all cases and they account for about 60% of CdLS; mutations in SMC1A and SMC3 account for a small percentage (Deardorff et al., 2011).
“The authors should compare prenatal findings in the current case with what described in the article by Clark et al. (2012). Without molecular testing data, I would like to see a table demonstrating the current case presenting prenatal clinical features consistent with what has been documented in the literature by studying a larger cohort of probands.”
Cornelia de Lange Syndrome (CdLS) is a multisystem developmental disorder characterized by growth retardation, cognitive impairment, external and internal structural malformations, and characteristic facial features. Currently, there are no definitive prenatal screening measures that lead to the diagnosis of CdLS. (Clark et al. 2012).
In our case (atypical CdLS) increased nuchal translucency in the first trimester, normal karyotype, saddle nose, micrognathia with receding jaw, low set ears, facies senilis, arthrogryposis of the hands, absence of the Aranzio ductus venous, dilatation of gallbladder and bowel, a unique umbilical artery, increased volume of amniotic fluid, and intrauterine growth retardation were found.
Fewer than 1% of individuals diagnosed with Cornelia de Lange syndrome have an affected parent.
Recommendations for the evaluation of parents of a proband with an apparent de novo mutation include clinical examination for features of CdLS, complete with plotting of growth parameters and molecular genetic testing if the NIPBL mutation has been identified in the proband. In our atypical Cornelia de Lange syndrome molecular tests of parents were negative.
“In the discussion, in the beginning of first paragraph, the authors claim that CdLS is diagnosed primarily by morphological characteristic and cited a paper published in 1933. At present, the diagnosis criteria of CdLS cannot be the same as what was 90 years ago, particularly with the identification of specific gene mutations in CdLS probands. Please re-phrase this part, and cite updated references.”
Diagnosis is based on clinical findings. High-resolution ultrasound examination to follow growth and to evaluate the limbs, heart, diaphragm, palate, and other organs or structures affected in CdLS is fundamental. Reported prenatal ultrasound findings include: increased nuchal translucency in the first trimester [Sekimoto et al., 2000; Huang & Porto, 2002]; Growth failure, which typically presents in the second trimester; and the typical in utero facial profile of a fetus with CdLS consisting of micrognathia, a prominent upper lip, and a depressed nasal bridge with somewhat anteverted nares [Ranzini et al., 1997; Boog et al., 1999; Urban & Hartung, 2001].
“In the end of the discussion, what does it mean “genetic tests are not reliable”? As a matter of fact, molecular testing has become a widely accepted concept and a popular diagnostic approach for individuals with congenital anormalies.”
Genetic tests are not reliable when negative. On the contrary when positive they are important for syndrome.
Indeed, mutations in NIPBL are not present in all cases and they account for about 60% of CdLS; mutations in SMC1A and SMC3 account for a small percentage (Deardorff et al., 2011).
Minor things:
These minor things are easy to correct. I thank the first referee for his clarifications.
Competing Interests:No competing interests were disclosed.Close
vito leanza, Surgery Department, Obstetrics and Gynecologic Unit “Ospedale Santo Bambino”, Catania University, Catania, Italy
01 Sep 2014
Author Response
“Data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) would be important for this case. The article published by Clark et al. (2012) which was
...
Continue reading
“Data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) would be important for this case. The article published by Clark et al. (2012) which was also cited by the authors, recommends molecular analysis of CdLS genes to prenatal diagnosis of CdLS.”
In our case report, data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) are negative , indeed it was called atypical Cornelia de Lange Syndrome (CdLS).
Mutations in NIPBL are not present in all cases and they account for about 60% of CdLS; mutations in SMC1A and SMC3 account for a small percentage (Deardorff et al., 2011).
“The authors should compare prenatal findings in the current case with what described in the article by Clark et al. (2012). Without molecular testing data, I would like to see a table demonstrating the current case presenting prenatal clinical features consistent with what has been documented in the literature by studying a larger cohort of probands.”
Cornelia de Lange Syndrome (CdLS) is a multisystem developmental disorder characterized by growth retardation, cognitive impairment, external and internal structural malformations, and characteristic facial features. Currently, there are no definitive prenatal screening measures that lead to the diagnosis of CdLS. (Clark et al. 2012).
In our case (atypical CdLS) increased nuchal translucency in the first trimester, normal karyotype, saddle nose, micrognathia with receding jaw, low set ears, facies senilis, arthrogryposis of the hands, absence of the Aranzio ductus venous, dilatation of gallbladder and bowel, a unique umbilical artery, increased volume of amniotic fluid, and intrauterine growth retardation were found.
Fewer than 1% of individuals diagnosed with Cornelia de Lange syndrome have an affected parent.
Recommendations for the evaluation of parents of a proband with an apparent de novo mutation include clinical examination for features of CdLS, complete with plotting of growth parameters and molecular genetic testing if the NIPBL mutation has been identified in the proband. In our atypical Cornelia de Lange syndrome molecular tests of parents were negative.
“In the discussion, in the beginning of first paragraph, the authors claim that CdLS is diagnosed primarily by morphological characteristic and cited a paper published in 1933. At present, the diagnosis criteria of CdLS cannot be the same as what was 90 years ago, particularly with the identification of specific gene mutations in CdLS probands. Please re-phrase this part, and cite updated references.”
Diagnosis is based on clinical findings. High-resolution ultrasound examination to follow growth and to evaluate the limbs, heart, diaphragm, palate, and other organs or structures affected in CdLS is fundamental. Reported prenatal ultrasound findings include: increased nuchal translucency in the first trimester [Sekimoto et al., 2000; Huang & Porto, 2002]; Growth failure, which typically presents in the second trimester; and the typical in utero facial profile of a fetus with CdLS consisting of micrognathia, a prominent upper lip, and a depressed nasal bridge with somewhat anteverted nares [Ranzini et al., 1997; Boog et al., 1999; Urban & Hartung, 2001].
“In the end of the discussion, what does it mean “genetic tests are not reliable”? As a matter of fact, molecular testing has become a widely accepted concept and a popular diagnostic approach for individuals with congenital anormalies.”
Genetic tests are not reliable when negative. On the contrary when positive they are important for syndrome.
Indeed, mutations in NIPBL are not present in all cases and they account for about 60% of CdLS; mutations in SMC1A and SMC3 account for a small percentage (Deardorff et al., 2011).
Minor things:
These minor things are easy to correct. I thank the first referee for his clarifications.
“Data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) would be important for this case. The article published by Clark et al. (2012) which was also cited by the authors, recommends molecular analysis of CdLS genes to prenatal diagnosis of CdLS.”
In our case report, data from mutational testing on known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) are negative , indeed it was called atypical Cornelia de Lange Syndrome (CdLS).
Mutations in NIPBL are not present in all cases and they account for about 60% of CdLS; mutations in SMC1A and SMC3 account for a small percentage (Deardorff et al., 2011).
“The authors should compare prenatal findings in the current case with what described in the article by Clark et al. (2012). Without molecular testing data, I would like to see a table demonstrating the current case presenting prenatal clinical features consistent with what has been documented in the literature by studying a larger cohort of probands.”
Cornelia de Lange Syndrome (CdLS) is a multisystem developmental disorder characterized by growth retardation, cognitive impairment, external and internal structural malformations, and characteristic facial features. Currently, there are no definitive prenatal screening measures that lead to the diagnosis of CdLS. (Clark et al. 2012).
In our case (atypical CdLS) increased nuchal translucency in the first trimester, normal karyotype, saddle nose, micrognathia with receding jaw, low set ears, facies senilis, arthrogryposis of the hands, absence of the Aranzio ductus venous, dilatation of gallbladder and bowel, a unique umbilical artery, increased volume of amniotic fluid, and intrauterine growth retardation were found.
Fewer than 1% of individuals diagnosed with Cornelia de Lange syndrome have an affected parent.
Recommendations for the evaluation of parents of a proband with an apparent de novo mutation include clinical examination for features of CdLS, complete with plotting of growth parameters and molecular genetic testing if the NIPBL mutation has been identified in the proband. In our atypical Cornelia de Lange syndrome molecular tests of parents were negative.
“In the discussion, in the beginning of first paragraph, the authors claim that CdLS is diagnosed primarily by morphological characteristic and cited a paper published in 1933. At present, the diagnosis criteria of CdLS cannot be the same as what was 90 years ago, particularly with the identification of specific gene mutations in CdLS probands. Please re-phrase this part, and cite updated references.”
Diagnosis is based on clinical findings. High-resolution ultrasound examination to follow growth and to evaluate the limbs, heart, diaphragm, palate, and other organs or structures affected in CdLS is fundamental. Reported prenatal ultrasound findings include: increased nuchal translucency in the first trimester [Sekimoto et al., 2000; Huang & Porto, 2002]; Growth failure, which typically presents in the second trimester; and the typical in utero facial profile of a fetus with CdLS consisting of micrognathia, a prominent upper lip, and a depressed nasal bridge with somewhat anteverted nares [Ranzini et al., 1997; Boog et al., 1999; Urban & Hartung, 2001].
“In the end of the discussion, what does it mean “genetic tests are not reliable”? As a matter of fact, molecular testing has become a widely accepted concept and a popular diagnostic approach for individuals with congenital anormalies.”
Genetic tests are not reliable when negative. On the contrary when positive they are important for syndrome.
Indeed, mutations in NIPBL are not present in all cases and they account for about 60% of CdLS; mutations in SMC1A and SMC3 account for a small percentage (Deardorff et al., 2011).
Minor things:
These minor things are easy to correct. I thank the first referee for his clarifications.
Competing Interests:No competing interests were disclosed.Close
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)