Keywords
Salmonids, Aquaculture, ecomorphs, Polymorphism, parallel evolution, immunology, craniofacial divergence, mtDNA
Salmonids, Aquaculture, ecomorphs, Polymorphism, parallel evolution, immunology, craniofacial divergence, mtDNA
Historical contingencies and chance shape organisms during evolution1,2, but convergence in phenotype and molecular systems indicates that evolution is to some extent predictable3,4. Identification of genes and variants that influence evolved differences is not a trivial task5. Ideal systems to study the role of chance and necessity in ecological evolution would be related species or populations with readily observable phenotypic variation, living in a tractable ecological setting, and most crucially showing parallel evolution of specific traits within/among species/populations. Examples of such a species complex are the finches of the Galapagos islands, cichlids in the African great lakes are exciting multi-species systems in this respect6,7. The threespine stickleback has also emerged as a model “single species” system8. The amount of diversity in the feeding specializations of fish provide great opportunities for studying adaptation and divergence at the developmental and genetic level.
Transcriptomic methods have been to address evolutionary and ecological questions in fish. For example microarrays were used to compare gene expression in anadromous and resident populations of brown trout (Salmo trutta), revealing that life history was a better predictor of gene expression in the liver than relatedness9. The newer technique, RNA-sequencing (RNA-seq) has been applied to species such as the Mexican cavefish (Astyanax mexicanus), cod (Gadus morhua) brook charr (Salvelinus fontinalis) and Atlantic salmon (Salmo salar)10–15, addressing questions concerning evolution, molecular genetics, development and aquaculture. RNA-seq was used to study salinity tolerance in Arctic charr, linking expression and quantitative trait loci16. Microarray studies of adult lake whitefish (Coregonus clupeaformis) pointed to parallel expression differences between benthic and limnetic forms17. Filteau et al. (2013)18 found that a set of coexpressed genes differentiated the two whitefish morphotypes, implicating Bone morphogenesis protein (BMP) signaling in the development of ecological differences in tropic morphology. One approach to identify pathways related to function or morphological differences is to study gene expression during development19,20.
Some northern freshwater fish species exhibit frequent parallelism in trophic structures and life history and in several cases are they found as distinct resource morphs8,21–25. One of these species, Arctic charr (Salvelinus alpinus), is well suited for studying the developmental underpinnings of trophic divergence and parallel evolution. Local adaptation has been extensively studied in the salmonid family, to which Arctic charr belongs26. The family is estimated to be between 63.2 and 58.1 million years old27,28. A whole genome duplication event occurred before the radiation of the salmonid family29–32 which has provided time for divergence of ohnologous genes (paralogous genes originated by whole genome duplication event). Furthermore, recent estimates from the rainbow trout (Oncorhynchus mykiss) genome suggest that ohnologous genes were lost at a rate of about 170 ohnologous genes per million years and by utilizing multiple data sources the genome assembly problem of this family can be solved32. De novo assembly of genomes and transcriptomes is complicated if many paralogs are present, such as in salmonids. Furthermore, for data with short reads, mapping to a related reference genome/transcriptome is recommended over de novo assembly33.
Following the end of the last glacial period, about 10.000 years ago, Arctic charr colonized northern freshwater systems34. It can be found as anadromous or lake/stream residents and exhibits high level of within species polymorphism23,34. Resource polymorphism in charr correlates with ecological attributes35–37. For instance small charr with benthic morphology, are found in multiple lavaspring and pond habitats in Iceland38, and a comparative study of Icelandic lakes37 found that lakes with greater limnetic habitat, fewer nutrients, and greater potential for zooplankton consumption appeared to promote resource polymorphism. Some of the larger lakes contain two or more distinct morphs, typically limnetic and benthic forms. Multiple lines of evidence show that these differences stem both from environmental and genetic causes39–43. The best studied example of sympatric charr are the four morphs in Lake Thingvallavatn44; two have a benthic morphotype, a large benthivorous (LB-charr) and a small benthivorous (SB-charr), and two morphs are limnetic, a large piscivorous morph (PI-charr) and small planktivorous morph (PL-charr)45. Both PL and PI-charr operate in open water and feed on free-swimming prey, PL on planktonic crustaceans and PI on small fish.
Several population genetics studies, using allozymes and mtDNA revealed no differences among charr populations46–48, while studies on microsatellite markers and nuclear genes, reveled both subtle49–51 and strong genetic differences among morphs52. Importantly Kapralova et al. (2011)51 concluded that small benthic morphs have evolved repeatedly in Iceland and that gene flow has been reduced between the PL and SB morphs in Lake Thingvallavatn since its formation approximately 10,000 years ago53. We also discovered genetic separation in immunological genes (MHCIIα and cath2) between morphs in Iceland and within the lake52, consistent with ecologically driven evolution of immune functions. Recently qPCR analyses showed that expression of mTOR pathway components in skeletal muscle correlates with the SB-charr form in Iceland54, but it is unknown whether there is genetic differentiation in those genes or upstream regulators.
Because individual genes have distinct histories55,56, genome wide methods are needed to identify genes and mutation that associate with divergence. Icelandic aquaculture charr (AC) was founded with fish from the north of Iceland, and has been bred at Holar University College since 199057. The Holar AC-charr has responded to artificial selection in growth and performance characteristics, and is now the dominant charr breed in aquaculture in Iceland. While clearly a derived form, it has retained general limnetic craniofacial morphotype (Figure 1). In this study we compare SB-charr from Lake Thingvallavatn and AC-charr because i) SB charr represents an extensively studied and derived form of charr, that has been separated from Anadromous fish for approx. 10,000 years, ii) of the availability of abundant AC material and iii) we wanted an extreme contrast, because of budget reasons we could only sequence 8 samples at the time. This rather extreme contrast is justified as the data and other studies58,59 building on this data illustrate (see discussion).

Adult individuals of the two morphs; the Holar aquaculture charr above and the small benthic charr from Lake Thingvallavatn below. Differences in size, coloration and head morphology are apparent.
The aims of this project are threefold. First, to find genes and pathways related to the development of phenotypic differences between benthic and limnetic Arctic charr morphs. Second, to screen for signals of genetic differentiation that may relate to divergence of benthic and limnetic charr. Third, we set out to verify a subset of the expression and genetic signals, in benthic and limnetic morphs. We conducted RNA-sequencing of developing offspring of two contrasting Arctic charr morphs, a small benthic charr from Lake Thingvallavatn and Icelandic aquaculture charr conforming to a limnetic morphotype. This identified candidate genetic changes and differential expression of developmental genes that may affect jaw and craniofacial traits which separate benthic and limnetic morphotypes in charr.
We set up crosses and reared embryos in the laboratory as previously described in 58. Embryos from four charr morphs were studied: an aquaculture charr (AC-charr) from the Holar breeding program57 and three natural morphs from Lake Thingvallavatn; small benthivorous (SB), large benthivorous (LB) and small planktivorous (PL) charr60. Samples of the first two, AC and SB-charr, which exhibit contrasting adult size and morphology (Figure 1), were collected in 2009 and material sent for developmental transcriptome sequencing. The latter two were sampled in 2010 and used for qPCR and SNP studies of selected genes. Briefly, in September 2009 we got material from spawning AC-charr from the Holar breeding program57 and spawning SB-charr collected via gill netting in Olafsdrattur in Lake Thingvallavatn. Similarly, in the 2010 spawning season SB-, LB- and PL-charr were collected from Lake Thingvallavatn. Fishing permissions were obtained from the Thingvellir National Park Commission and the owner of the Mjóanes farm. For each parent group, eggs from several females were pooled and fertilized using milt from several males from the same group. Embryos were reared at ~5°C under constant water flow and in complete darkness at the Holar University College experimental facilities in Verid, Saudárkrókur. The water temperature was recorded twice daily and the average was used to estimate the relative age of the embryos using tausomite units (τs)61. Embryos and juveniles were sampled at designated time points, placed in RNAlater (Ambion) and frozen at -20°C. Post hatching juveniles were reared at the same temperature on standard Aquaculture food. For the investigation of different tissues of adult aquaculture charr (AC) from Hólar (fish size 20–25 cm) were used. Six randomly selected individuals were killed (by cutting through spinal cord) and dissected, and samples were taken from the skin, heart, liver, gills, spleen, intestine and kidney of each fish. The samples were placed in RNAlater (Ambion) and stored at -20°C. DNA for population genetic analyses was taken from our previous study52.
Fishing in Lake Thingvallavatn was with permissions obtained both from the owner of the land in Mjóanes and from the Thingvellir National Park commission. Ethics committee approval is not needed for regular or scientific fishing in Iceland (The Icelandic law on Animal protection, Law 15/1994, last updated with Law 157/2012). Sampling was performed by Holar University College Aquaculture Research Station (HUC-ARC) personnel. HUC-ARC has an operational license according to Icelandic law on aquaculture (Law 71/2008), which includes clauses of best practices for animal care and experiments.
Embryos of AC- and SB-charr sampled in 2009 were used for transcriptome sequencing. For this we focused on the time covering development of pharyngeal arches and morphogenesis of the head: at 141, 163, 200 and 433 τs (post fertilization). For each combination of morphs and timepoints we pooled RNA from approximately six individuals. RNA extraction and following steps were performed as described earlier58,62. The embryos were dechorionated and homogenized with a disposable Pellet Pestle Cordless Motor tissue grinder (Kimble Kontes, Vineland, NJ, USA) and RNA was extracted into two size-fractions using the Ambion mirVana kit (Life Technologies, Carlsbad, CA, USA). The high molecular weight fraction was further used for mRNA-seq and RNA quality was analysed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). First and second strand cDNA synthesis, fragmentation, adapter ligation and amplification were performed using the mRNA-Seq 8-Sample Prep Kit (Illumina, San Diego, CA, USA) according to manufacturer’s instructions. Sequencing was performed at DeCode genetics (Reykjavík, Iceland) using SOLEXA GAII technology (Illumina, San Diego, CA, USA). The sequencing reads were deposited into the NCBI SRA archive under BioProject identifier PRJNA239766 and with accession numbers: SRX761559, SRX761571, SRX761575, SRX761577, SRX761451, SRX761461, SRX761490 and SRX761501.
The embryos sampled in 2010 were used for qPCR expression analyses. RNA was extracted from six whole embryos, in two replicates (two repetitions X three fish) (AC and SB sampled at 161 and 200 τs). For the extraction of RNA from heads of AC, SB, LB and PL, 12 embryos (two repetitions X six fish) at 178, 200 and 216 τs were used. Embryos were dechorionated and decapitated in front of the pectoral fin. RNA extraction and cDNA preparation were performed as described previously in 58. Similarly, RNA was extracted from a small piece (approximately 2 mm2) of skin, heart, liver, gill, spleen, intestine and liver from six adult AC-charr.
As no S. alpinus genome is available and de novo assembly of the 36 bp reads yielded an excessive number of short contigs we chose to assess expression and genetic variation by mapping the reads to 59336 S. salar expressed sequence tag (EST) contigs from the SalmonDB [63, downloaded 22. March 2012] and the Arctic charr mitochondrial genome [55, NC_000861].
To estimate expression, reads were aligned with RSEM version 1.1.18 with default parameters. RSEM distributes reads that map to multiple locations to the most likely contig, using expectation maximization64. The read counts for contigs with the same annotation were pooled because some genes were represented by more than one contig, and due to whole genome duplication salmonids have closely related paralogous genes30,32. Thus the expression tests are done on gene or paralog group level, instead of the contig level. In the remainder of the paper, the term ’gene’ will have this broader meaning, some genes are represented by one contig and others by two or more (indicated in all relevant tables). This brought the number of genes considered down to 16851. Lastly, genes with fewer than 800 mapped reads in the entire dataset were excluded from the analyses, yielding a total of 10496 genes.
A generalized linear model (GLM) with morph and developmental time as explanatory variables was used to find genes with different expression levels between the two charr morphotypes (groups) using the edgeR-package in R65.
Y = Morph + Time + Error
We could not test for interaction, as biological replicates were unavailable. To obtain further insight into the expression profiles of differently expressed genes, we preformed clustering analyses on log-transformed cpm-values (counts per million; cpm-function in edgeR). The values for each gene were scaled by mean and standard deviation, and the euclidean distance used for the hclust-function in R66 with the default settings. We used the hypergeometric-test in goseq67 to test for gene ontology enrichment. Since we pooled the read-count from different contigs we could unfortunately not take gene length into account in those tests.
We previously identified suitable reference genes to study Arctic charr development58. Here we examined the expression of several genes in whole charr embryos, embryonic heads and adult tissues. Primers were designed using the Primer3 tool68 and checked for self-annealing and heterodimers according to the MIQE guidelines69 (S1 Table). Primers for genes with several paralogs were designed for regions conserved among paralogs. For natterin, primers for the different paralogs were designed to match regions differing in sequence. Relative expression was calculated using the 2−∆∆Ct method70. For the calculation of relative expression of genes in whole embryos, the geometric mean expression of three reference genes, β-Actin, elongation factor 1α and Ubiquitin-conjugating enzyme E2 L3, was used for normalization. For visual comparisons among samples, the normalized expression was presented as relative to the expression in AC at 161 τs (calibration sample). For the embryonic head samples IF5A1 and ACTB were used as reference genes and a biological replicate of AC at 178 (τs) as the calibrator sample. Standard errors of relative expression were calculated from the standard errors (SE) of the ∆CT-values with the formula 2-(∆∆Ct+SE) = minimum fold expression and 2-(∆∆Ct−SE) = maximum fold expression. The statistical analysis was performed using the ∆CT-values with a two-way ANOVA with GLM function in R.
Y = Morph + Time + M x T + Error
Normal distribution of residuals was confirmed for all data. For the study of expression in the embryonic head we followed a significant morph effect in the ANOVA with Tukey’s post-hoc honest significant difference test, on relative expression ratios (∆CTs).
For analysis of genetic variation we mapped the reads to the salmon contigs, this time using the Burrows-Wheeler Aligner (BWA)71 with a seed length of 25, allowing two mismatches. We re-mapped the reads, since BWA allows short indels (RSEM does not) but disregarding them leads to many false SNPs close to indels. To extract candidate polymorphic sites from the Arctic charr transcriptome we ran VarScan272 with minimum coverage of 50 reads and minimum minor allele frequency of 0.1 on reads mapped to each S. salar contig for all of the 8 timepoints and morph combinations. This was done separately for reads that mapped uniquely to one contig only (UNI) and reads that mapped to two or more contigs (REP). These SNP-candidates were further processed in R66. SNP-candidates at 90% frequency or higher in all samples were disregarded, as they reflect differences between Arctic charr and S. salar and are not the focus of this study. SNP-candidates with poor coverage in specific samples - i.e. coverage of five or fewer reads in three or four samples of each morph - were removed. As the SNP analysis was done on individual contigs, differences among paralogs appear in the data. However, since each sample is a pool of few individuals, it is very unlikely that we have the same frequency of true SNPs in the samples. This property was used to remove variants that are most likely due to expressed paralogs. Using Fisher exact tests to evaluate differences between samples, only SNPs that were significantly different between samples with a p < 0.05 (with no multiple testing correction) were chosen for further examination. As equal cDNA input from individuals in sample cannot be assumed, due to expression differences among them and stochastic processes in sample preparation, read numbers were summed over the four samples from each morph for the comparison between them. A conservative approach was taken to look for differences between morphs. We focused on SNP-candidates that showed differences in frequency between morphs, without adjusting for multiple testing (Fisher exact test, p > 5%). We extracted the most interesting candidates by filtering by frequency difference between the morphs (delta). SNP-candidates with the highest frequency difference (delta > 95%) were manually processed and redundant candidates removed. A similar approach was used to mine for polymorphisms in Arctic charr mtDNA (NC_000861), using S. salar mtDNA as the outgroup (NC_001960.1).
We wrote a python script to predict the impact of SNPs within the mRNA sequences. Polymorphisms were categorized according to their location (3’UTR, coding, 5’UTR), and those within the coding region into synonymous or non-synonymous.
We chose 12 candidate SNPs for verification (see below). The candidates were verified using a similar approach as previously52; first, we conducted genomic comparisons of the Salmon genome, ESTs and short contigs from the preliminary assembly of the Arctic charr transcriptome. This allowed us to infer the placement of the putative polymorphism in the locus, and design paralog specific primers for PCR (less than 1 kb amplicons) for verification of the 12 candidate SNPs (S2 Table). MJ tetrad machine was used for PCR and the program was 5 min. at 95°C, followed by 35 cycles of 30 sec. at 52°C, 1 min. at 72°C, 30 sec. at 95°C, ending with 12°C while waiting on the human. Each individual was genotyped by first amplifying the region of interest using PCR, followed by ExoSAP (Affymetrix), direct sequencing (BigDye) and finally run on an Applied Biosystems 3500xL Genetic Analyzer (Hitachi). Raw data was base-called using the Sequencing Analysis Software v5.4 with KBTMBasecaller v1.41 (Applied Biosystems). Ab1 files were run through Phred and Phrap and imported to Consed for visual editing of ambiguous bases and putative polymorphisms, and for trimming primers. The FASTA files were aligned with ClustalW online [73, http://www.ebi.ac.uk/Tools/msa/clustalw2/] and manually inspected in Genedoc74. All sequences where deposited to Genbank as popsets under the accession numbers KP019972-KP020026.
Comparative genomics showed that several verified SNPs affected evolutionarily constrained parts of the mitochondrial genome. Two approaches were used: performing a BLAST search on salmon ESTs (May 2013) and retrieving multiZ alignments of vertebrates from the UCSC genome browser (in September 2013). This yielded several hundred sequences from related fish and other vertebrates. The list was reduced to 20 sequences for visualization, by keeping members of the major taxa but removing more closely related sequences, aligned with ClustalW and manually adjusted in Genedoc. The species and genome versions used are; Human (Homo sapiens, hg19), Lamprey (Petromyzon marinus, petMar1), Fugu (Takifugu rubripes, fr2), Medaka (Oryzias latipes, oryLat2), Stickleback (Gasterosteus aculeatus, gasAcu1), Tetraodon (Tetraodon nigroviridis, tetNig2), Zebrafish (Danio rerio, danRer6). We also downloaded from NCBI the sequence of whole or partial mtDNA from several fish species; Brown trout (Salmo trutta, JQ390057 and AF148843), Broad whitefish (Coregonus nasus, JQ390058), Legless searsid (Platytroctes apus, AP004107), Pacific menhaden (Ethmidium maculatum, AP011602), Icefish (Salanx ariakensis, AP006231 and HM151535), Chain pickerel (Esox niger, AP013046) and Western Pacific roughy (Hoplostethus japonicus, AP002938). The three mitochondrial variants (numbered by the S. alpinus mtDNA - NC_000861) are; m1829G>A (CCACGTTGTGAAACCAAC[G/A]TCCGAAGGTGGATTTAGCAGT), m3211T>C (CGTGCAGAAGCGGGCATAAG[T/C]ACATAAGACGAGAAGACCCT) and m3411C>T (CTCTAAGCACCAGAATTT[C/T]TGACCAAAAATGATCCGGC).
Each sample yielded good quality data, with sequencing depth from 49 to 58 million (average: 55 million) reads. To quantify the expression levels, the reads were aligned to a salmon EST-assembly63. Around 20% of the reads mapped uniquely to the EST data (S3 Table). A further 30% mapped to two or more contigs, probably representing paralogous genes, recent duplications or repeat-like elements within transcribed regions. A substantial fraction of the RNA-sequencing reads did not map to the contigs from S. salar. Analyses of those reads require an Arctic charr genome sequence or transcriptome assembly from longer and paired end reads.
For the expression analysis, ESTs were collapsed into 16851 “genes” or paralog groups (see the Methods for the broader meaning of gene). We only considered genes (total of 10496) with 800 or more reads mapped and tested for differential expression using the edgeR-package65. We detected considerable changes in the transcriptome during Arctic charr development (Figure 2a). The expression of 1603 and 2459 genes differed significantly between developmental timepoints at the 1% and 5% levels of false discovery rate (FDR), respectively (S1 file). The difference was most pronounced between pre-hatching (timepoints: 141, 163, 200 τs) and post hatching embryos (timepoint 433 τs), as more than 70% of the genes with FDR below 1% had higher expression in the latter (Figure 2a). According to Gene Ontology analyses, six separate GO categories are significant (below 10% FDR). The most drastic changes were seen in processes related to glycolysis (GO:0006096, FDR = 0.0009), were the expression of 19 out of 25 genes changed during this developmental period. The other five classes that were differentially expressed during charr development are: ion transport (GO:0006811, FDR = 0.027), blood coagulation (GO:0007596, FDR = 0.03), DNA repair (GO:0006281, FDR = 0.08) and two immune related categories (GO:0019882, FDR = 0.08, GO:0006955, FDR = 0.09). Those results probably reflect developmental changes and/or differences in the environment of embryos before and after hatching.
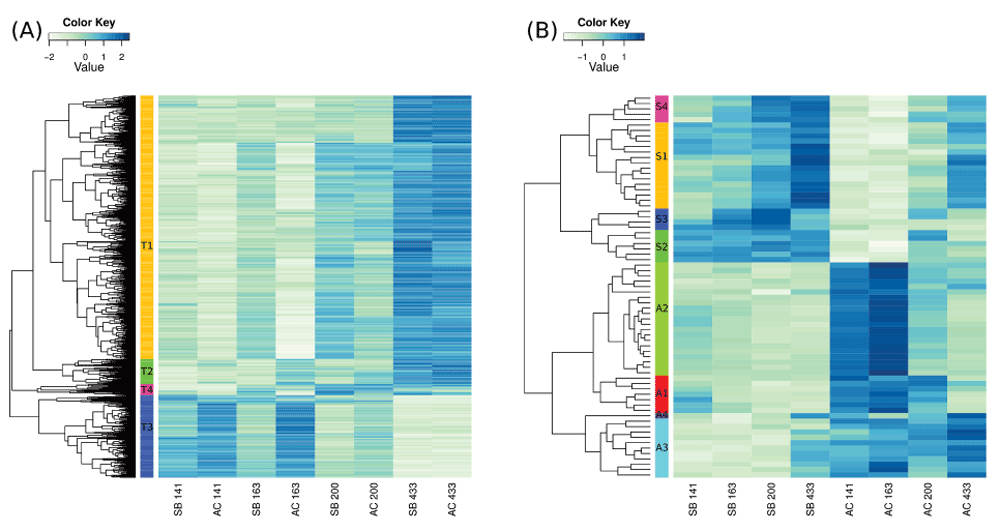
Two morphs (SB and AC) are represented, at four timepoints. (A) The 1603 genes with expression difference among time points, here clustered into four groups . (B) The 71 genes differentially expressed between morphs, are clustered into 4 groups for each of the two morphs. High expression is indicated by blue and low expression by beige.
We were especially interested in genes showing expression differences between the two Arctic charr morphs as they might implicate pathways involved in the ecological divergence among charr populations. In the data 296 genes were differentially expressed (FDR < 5%) between the morphs (141 higher in SB and 152 higher in AC, S1 file). Among genes with higher expression in SB-charr two biological GO categories were enriched: blood coagulation (GO:0007596, p = 0.001) and proteolysis (GO:0006508, p = 0.002). Recall, expression of blood coagulation factors also differed between developmental stages (see above). In AC-charr, genes in three categories: respiratory electron transport chain (GO:0022904, p = 0.0006), ATP synthesis coupled electron transport (GO:0042773, p = 0.002) and neurotransmitter transport (GO:0006836, p = 0.009) have higher expression. The first two GO categories both relate to energy generation in mitochondria and could reflect higher expression of genes with mitochondrial functions in AC-charr.
At more stringent FDR (1%), 31 genes were higher expressed in SB and 40 genes higher in AC-charr (Figure 2b, Table 1 and Table 2). These genes have diverse functional annotations. The genes with higher expression in each morph were clustered into 4 groups, which aggregated genes of similar function. For instance SB cluster 3 has three immune related genes: Complement factor D (9), H-2 class I histocompatibility antigen L-D alpha chain (2) and Sushi domain-containing protein 2 (4) and one gene with unknown function (Table 1). Note, however, that immune genes were not significantly enriched in the GO comparison of morphs.
The cluster numbering corresponds to Figure 1.
For column header explanation, see footer of Table 1.
The results suggest mitochondrial function and blood coagulation genes are differentially expressed between the morphs, but due to few samples used in the RNA-sequencing, qPCR verification was needed.
The data highlights genes likely to differ in expression between embryos of SB and AC-charr. Of the nine genes subjected to qPCR analyses of whole embryos, five were confirmed to be differentially expressed between AC and SB at 161 or 200 τs (Figure 3, S4 Table and S2 file). Three genes, Nattl, Alkaline phosphatase (Alp) and Lysozyme (Lyz), had significantly higher expression in SB. The other two, Keratin-associated protein 4–3 (Krtap4–3) and Poly polymerase 6 (Parp6) had higher expression in AC embryos (Figure 3, S4 Table). No morph and time interaction was detected for any of the genes.
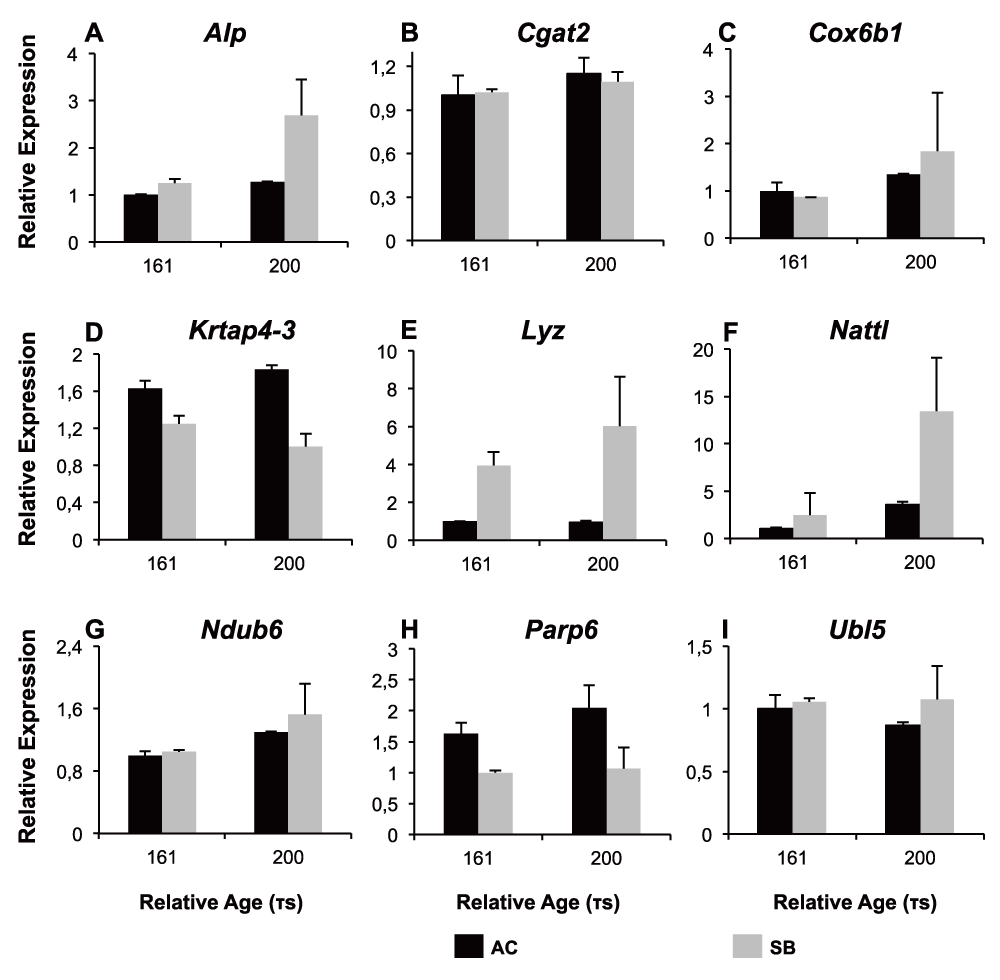
Relative expression of 9 genes (A–I) analysed by qPCR in the small benthic (SB) charr from Lake Thingvallavatn and aquaculture (AC) charr at two different developmental timepoints (161 and 200 $\tau s$). 5 genes were differentially expressed between the two morphs (Alp, Krtap4-3, Lyz, Nattl, Parp6), while 4 further genes did not show significant expression differences between morphs (Cgat2, Cox6B1, Ndub6, Ubl5), see Table S3. Error bars represent standard deviation calculated from two biological replicates.
As some of the genes are represented by different contigs or even paralogs, we set out to disentangle the expression of one paralog group in detail. The qPCR primers used above matched conserved gene regions and thus estimate the combined expression of several paralogs. We chose to measure the expression of three different natterin paralogs (nattl1, 2 and 3), in part because this understudied gene was first characterized as a toxin produced by a tropical fish75,76. We studied nattl expression in several developmental stages in AC-, SB- and PL-charr as well as in selected tissues of adult AC-charr. The expression level of the three paralogs differed between morphs and timepoints (Figure 4 and S5 Table). Overall nattl2 had the highest expression in all morphs. The nattl1 had higher expression in embryos of PL-charr than in AC- and SB-charr, while nattl2 and nattl3 were more expressed in SB-embryos.
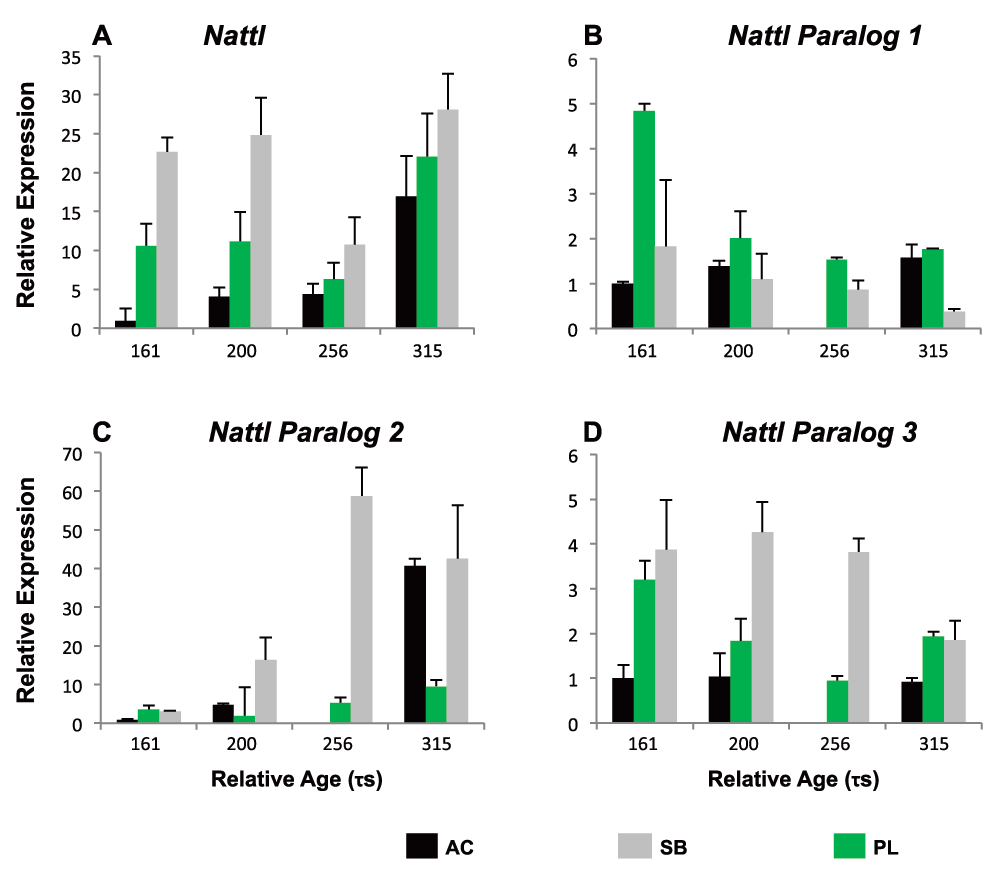
The expression is graphed for different morphs (SB, AC and PL) at four developmental timepoints (161, 200, 256 & 315 $\tau s$, relative to AC-charr at timepoint 161. A) General {Nattl} expression along charr development. B–D) Expression of Nattl paralogs 1–3. ANOVA showing the variation among morphs is summarized in Table S4.
In order to evaluate the hypothesis that nattl genes have immune-related functions we studied expression in adult tissues (in AC-charr). The nattl expression was highest in the gills, followed by expression in kidney, skin and spleen. Low expression levels were detected in liver, intestine and heart (S1 Figure and S5 Table). The three nattl paralogs followed different patterns, whilst each of them showed significant expression differences among tissues. Nattl1 was mainly expressed in spleen and kidney, while nattl2 showed a significantly higher expression in skin, liver and in gills. Similarly, the relative expression of nattl3 was highest in the gills and skin. This indicates that the three nattl paralogs are expressed in a tissue specific manner, and also differently during the development of the three charr morphs studied here.
To study the craniofacial divergence between sympatric Arctic charr morphs we used qPCR to study 8 genes with expression difference in the RNA-seq data (all higher in SB). We focused on genes with known craniofacial expression in zebrafish development77 and compared two benthic (SB, LB) and two limnetic charr (AC, PL). We analyzed heads at three time-points (178, 200 and 218 τs) as this period overlaps with early stages of craniofacial skeletal formation in Arctic charr78,79. The qPCR confirmed the higher expression of seven out of these eight genes in the head of benthic charr compared to limnetic charr (Figure 5, S2 Figure and S3 file). These seven genes are Claudin 4 (Cldn4), adseverin (Scin), Junction plakoglobin (Jup), Lipolysis stimulated lipoprotein receptor (Lsr), Major vault protein (Mvp), Transforming growth factor beta receptor II (Tgfbr2) and Vitamin D receptor a (Vdra). The eighth gene, Retinoic acid receptor gamma-A (Rarg) gave a small but significant response in the head, but the effects were reversed, i.e. the expression was higher in AC. The expression difference of the seven genes was, in almost all cases, consistent over the three time-points studied (See S2 Figure).
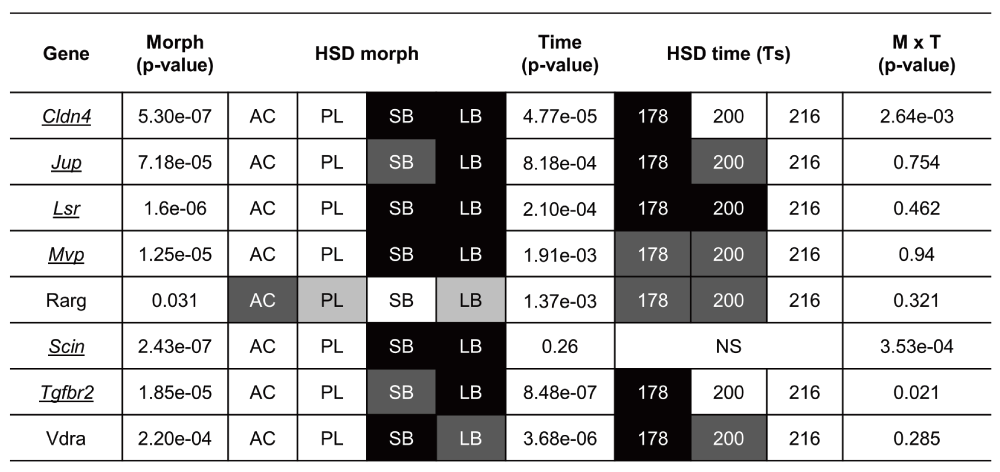
Relative expression ratios, calculated from the qPCR data, were subjected to an ANOVA to test the expression differences amongst four charr groups and three close time points ($\tau s$). The underlined gene names reflect significant difference between SB and AC-charr. A post hoc Tukey's test (HSD) was performed to determine the effects of morphs, time and morph-time interaction (M X T). White boxes represent low expression, while black boxes represent high expression. The shading represents significant different expression between the samples (p = 0.05, NS = not significant).
The RNA-seq data also revealed segregating variations with large frequency differences between charr morphs. To uncover candidate SNPs we mapped the reads to all of the S. salar EST-contigs. Filtering on coverage yielded 165,790 candidate SNPs (Table 3); of those 66.569 came from reads that mapped uniquely and 57.009 candidate SNPs from reads that mapped to more than one contig; with limited overlap between lists. Assuming that the expression of paralogous genes is stable, then differences among paralogs appear as SNPs at similar frequency in all samples. By requiring variant frequency differences (p < 0.05, uncorrected) between samples we reduced the list of candidates by two thirds, yielding over 20.000 candidate SNPs. Note, that as cDNA from charr families was sequenced (not a population sample), estimates of SNP frequencies are imprecise. To err on the side of caution, we chose SNP candidates with 50% or higher frequency difference between morphs for further study. The candidate SNPs were also summarized by frequency of the derived allele, in reference to the S. salar sequence. This gave 672 and 872 SNPs at higher frequency, in AC-charr and SB-charr, respectively. The uniquely and multiply mapped reads, revealed approximately similar numbers of candidate SNPs. Gene ontology analysis showed that for derived SNPs in SB, there was an excess of variants in genes related to translation, both as a broad category and specific subgroups (S6 Table). There was also enrichment of SNPs in genes related to DNA-mediated transposition, DNA integration, DNA replication and oxidation-reduction process. No GO categories were enriched for high frequency derived SNPs in AC. Furthermore, functional effects of the candidate SNPs (UTR, synonymous and non-synonymous) were predicted. The distribution among those categories did not differ between variants detected by uniquely or repeatedly mapped reads, χ2[3] = 2.59, p = 0.46 (S7 Table).
For Delta > 0.95 we show the number of SNP-candidates before the redundant ones were removed.
A total of 60 candidate SNPs are nearly fixed in one morph, with frequency difference between morphs above 95% (after manual inspection of contigs and SNP position three candidates were removed since they represented the same SNP). Of these “fixed” SNPs 46 came from uniquely mapped reads and 14 from reads that mapped more than twice (Table 4 and Table 5). For the SNPs from uniquely mapped reads, 17 are fixed in AC-charr and 29 in SB-charr. The few genes with two or more polymorphic sites were; Keratin type II cytoskeletal 3 (KRT3), Cysteine sulfinic acid decarboxylase (CSAD) and DNA-directed RNA polymerase I subunit RPA12 (RPA12) with 5, 5 and 2 SNPs respectively. KRT3 and CSAD had significant differentiation in both SB and AC. Similarly, 14 SNPs with large differentiation between morphs were predicted from reads that mapped on two or more contigs (Table 5). Of these, we found two variants in the mitochondrial 60S ribosomal protein L36 (RPL36) and variants in 4 other mitochondrial genes (28S ribosomal protein S18a mitochondrial (MRPS18A), Apoptosis-inducing factor 1 mitochondrial (AIFM1), Isocitrate dehydrogenase [NADP] mitochondrial (acIDH1) and Protein S100-A1 (S100A1)), all at higher frequency in AC-charr. PCR and Sanger sequencing was used to confirm SNPs in DNA2-like helicase (DNA2), a gene with nuclear and mitochondrial function, and two other genes Uroporphyrinogen decarboxylase (UROD), and Mid1-interacting protein 1-like (MID1IP1) (S2 Table). The candidate variant Eukaryotic translation initiation factor 4 gamma 2 (EIF4G2) was not substantiated by the PCR/sequencing.
| (a) Higher frequency in AC morph | |||||||
|---|---|---|---|---|---|---|---|
| Contig | Annotation | Pos | Ref | Var | Freq- SB | Freq- AC | Effect |
| SS2U026955 | Keratin type II cytoskeletal 3 | 300 | A | T | 0.000 | 0.984 | synonymous |
| SS2U026955 | Keratin type II cytoskeletal 3 | 309 | G | A | 0.000 | 0.996 | synonymous |
| SS2U033960 | Cysteine sulfinic acid decarboxylase | 192 | C | G | 0.000 | 1.000 | 5prime |
| SS2U033960 | Cysteine sulfinic acid decarboxylase | 416 | G | T | 0.000 | 0.961 | G to V |
| SS2U033960 | Cysteine sulfinic acid decarboxylase | 945 | C | A | 0.004 | 0.956 | synonymous |
| SS2U043396 | Eukaryotic translation initiation factor 2-alpha kinase 1 | 134 | A | G | 0.000 | 1.000 | 5prime |
| SS2U043886 | Transcription cofactor HES-6 | 1308 | T | C | 0.000 | 1.000 | 5prime |
| SS2U044339 | Intraflagellar transport protein 52 homolog | 479 | T | C | 0.021 | 1.000 | D to G |
| SS2U045168 | Putative Peptide prediction | 1275 | G | A | 0.000 | 1.000 | 3prime |
| SS2U045328 | E3 ubiquitin-protein ligase DTX3L | 388 | G | A | 0.000 | 0.977 | synonymous |
| SS2U045990 | Low-density lipoprotein receptor-related protein 1 | 135 | T | C | 0.000 | 0.969 | synonymous |
| SS2U048125a | Transmembrane protein 131-like | 480 | G | A | 0.000 | 1.000 | synonymous |
| SS2U052747 | Uridine 5’-monophosphate synthase | 914 | G | A | 0.000 | 0.951 | synonymous |
| SS2U054542 | Mediator of RNA polymerase II transcription subunit 20 | 474 | C | T | 0.027 | 0.995 | synonymous |
| SS2U056193 | SUMO-conjugating enzyme UBC9 | 96 | A | T | 0.000 | 1.000 | 3prime |
| SS2U057101 | ETS domain-containing protein Elk-3 | 440 | C | G | 0.000 | 1.000 | 3prime |
| SS2U058860 | Voltage-dependent anion-selective channel protein 2 | 681 | G | T | 0.000 | 1.000 | 3prime |
| (b) Higher frequency in SB morph | |||||||
| SS2U000399 | Insulin-like growth factor-binding protein 7 | 598 | C | A | 1.000 | 0.000 | 3prime |
| SS2U004484 | Titin | 387 | G | A | 0.990 | 0.010 | synonymous |
| SS2U026826 | L-asparaginase | 363 | C | T | 1.000 | 0.000 | H to Y |
| SS2U026955 | Keratin type II cytoskeletal 3 | 116 | C | A | 0.996 | 0.031 | T to N |
| SS2U026955 | Keratin type II cytoskeletal 3 | 264 | C | T | 0.970 | 0.008 | synonymous |
| SS2U026955 | Keratin type II cytoskeletal 3 | 317 | C | T | 1.000 | 0.002 | T to M |
| SS2U033960 | Cysteine sulfinic acid decarboxylase | 363 | C | T | 1.000 | 0.025 | 5prime |
| SS2U033960 | Cysteine sulfinic acid decarboxylase | 387 | C | T | 1.000 | 0.030 | synonymous |
| SS2U033960 | Cysteine sulfinic acid decarboxylase | 657 | T | C | 0.990 | 0.031 | synonymous |
| SS2U034322 | Cyclin-C | 1094 | A | G | 1.000 | 0.000 | 3prime |
| SS2U034431 | Dolichyl-diphosphooligosaccharide–protein glycosyltransferase subunit 2 | 436 | G | A | 0.992 | 0.000 | G to S |
| SS2U036025 | Nuclear receptor coactivator 4 | 36 | G | A | 1.000 | 0.043 | 5prime |
| SS2U040590 | Glutamyl-tRNA(Gln) amidotransferase subunit A homolog | 478 | G | A | 0.972 | 0.000 | synonymous |
| SS2U045606 | Superkiller viralicidic activity 2-like 2 | 500 | C | T | 1.000 | 0.000 | synonymous |
| SS2U047816 | Squalene synthase | 1139 | G | A | 1.000 | 0.029 | synonymous |
| SS2U048063 | Lysine-specific demethylase NO66 | 669 | C | T | 1.000 | 0.000 | synonymous |
| SS2U050394 | UPF0542 protein C5orf43 homolog | 596 | G | A | 1.000 | 0.000 | synonymous |
| SS2U050880a | Transmembrane protein 131-like | 901 | C | T | 1.000 | 0.000 | A to V |
| SS2U052076 | Eukaryotic translation initiation factor 3 subunit A | 824 | C | T | 1.000 | 0.031 | synonymous |
| SS2U053417 | RNA polymerase-associated protein LEO1 | 454 | G | A | 1.000 | 0.049 | synonymous |
| SS2U054333 | Scaffold attachment factor B2 | 382 | G | A | 0.999 | 0.000 | V to M |
| SS2U054705 | Cell division protein kinase 4 | 122 | A | G | 0.971 | 0.000 | 3prime |
| SS2U054965 | DNA-directed RNA polymerase I subunit RPA12 | 106 | G | A | 1.000 | 0.000 | 5prime |
| SS2U054965 | DNA-directed RNA polymerase I subunit RPA12 | 411 | T | G | 1.000 | 0.000 | synonymous |
| SS2U055120 | Chromatin modification-related protein MEAF6 | 350 | A | C | 1.000 | 0.000 | H to P |
| SS2U055153 | Complexin-1 | 1191 | C | A | 1.000 | 0.031 | 3prime |
| SS2U057635 | Mitogen-activated protein kinase 14B | 1370 | A | T | 1.000 | 0.026 | 3prime |
| SS2U058169 | Transmembrane protein 50A | 1214 | C | G | 0.973 | 0.000 | 3prime |
| SS2U058802 | Signal recognition particle 54 kDa protein | 607 | T | A | 0.969 | 0.000 | C to S |
| Contig | Annotation | Pos | Ref | Var | Freq- SB | Freq- AC | Effect |
|---|---|---|---|---|---|---|---|
| SS2U004839 | Actin alpha sarcomeric/cardiac | 550 | A | C | 0.015 | 0.999 | 3prime |
| SS2U021298 | 28S ribosomal protein S18a mitochondrial | 462 | A | C | 0.000 | 1.000 | synonymous |
| SS2U041264 | Apoptosis-inducing factor 1 mitochondrial | 341 | C | T | 0.000 | 0.952 | synonymous |
| SS2U054211a | Cytoplasmic dynein 1 intermediate chain 2 | 136 | T | C | 0.018 | 0.974 | synonymous |
| SS2U054362a | Q08CA8 Dynein cytoplasmic 1 intermediate chain 2 | 945 | A | G | 0.000 | 1.000 | synonymous |
| SS2U055923 | Bystin | 1623 | A | C | 0.000 | 0.983 | 3prime |
| SS2U058758 | Protein S100-A1 | 253 | C | T | 0.000 | 0.984 | synonymous |
| SS2U059000 | Isocitrate dehydrogenase [NADP] mitochondrial | 1654 | T | C | 0.000 | 0.975 | 3prime |
| SS2U059146 | 60S ribosomal protein L36 | 263 | T | G | 0.009 | 1.000 | synonymous |
| SS2U059146 | 60S ribosomal protein L36 | 470 | A | C | 0.009 | 1.000 | synonymous |
| SS2U036667 | Heterogeneous nuclear ribonucleoprotein K | 813 | C | T | 1.000 | 0.022 | 5prime |
| SS2U042873 | RNA polymerase-associated protein LEO1 | 460 | G | A | 1.000 | 0.000 | synonymous |
| SS2U058455 | Adenylosuccinate lyase | 1616 | C | T | 1.000 | 0.000 | 3prime |
| SS2U058906 | Mid1-interacting protein 1-like | 350 | G | T | 0.985 | 0.000 | E to D |
Considering the enrichment of differentially expressed genes related to mitochondrial energy metabolism (above), and high frequency candidate SNPs in several genes with mitochondrial function in AC-charr we decided to study the mitochondrial transcriptome further. The charr studied here reflect metabolic extremes, the aquaculture charr was bred for growth while the small benthic morph is thought to have experienced natural selection for slow metabolism and retarded growth45,80. Although mRNA preparation protocols were used for generating cDNA for the RNA-sequencing, a substantial number of reads came from non-polyadenylated sequences. By mapping the reads to mtDNA sequence of Arctic charr we could estimate expression and infer polymorphism both in genes and intergenic regions. There was a clear difference in sequencing coverage, with more than twice as many reads mapped from the AC- compared to SB-charr (mean fold difference 2.27, Wilcoxon test, p < 0.0004). Note, as only two types of fish are compared, it is impossible to determine the polarity of expression divergence.
The mapped RNA-reads were used to identify polymorphism and divergence in the entire mitochondrial chromosome. The polymorphisms were found by mapping to mtDNA from a Canadian S. alpinus55, but ancestral vs. derived status inferred by comparison to S. salar mtDNA. Bioinformatics revealed 82 candidate sites, including 35 that represent divergence between Icelandic and Canadian charr. A total of 20 candidate SNPs had high (more than 50%) frequency difference between SB and AC-charr (Figure 6). There was no bias in the distribution of derived SNPs, 11 on the AC branch and 9 in SB. Note, the frequency distribution is most irregular as we sequenced embryos of related individuals (see Materials and Methods), not a population sample. The divergence between Iceland and Canada is particularly little in the 12s and 16s ribosomal RNA genes. Curiously in those genes were two SNPs differing strongly in frequency between morphs (Figure 6). To confirm and better estimate the frequency of variants in the ribosomal genes, we PCR amplified and sequenced two ~550 bp regions in the rRNA genes, comparing three morphs (PL, LB and SB) from Lake Thingvallavatn (Figure 7A, C and E, S2 Table). The 12s polymorphism (m1829G>A) differed significantly between the morphs (χ2[2] = 8.6, p = 0.014), and was at highest frequency in the SB (0% in PL, 12.5% in LB and 75% in SB). Similarly m3411C>T in the 16s was enriched in SB (62.5%) but found at lower frequency in PL (0%) and LB (12.5%) (it differed significantly between morphs, χ2[2] = 9.3333, p = 0.009). The Sanger sequencing also revealed three other polymorphisms in the amplified region, not seen in the transcriptome. Among those m3211T>C in the 16s gene was at 75% frequency in LB, but not found in the other morphs (χ2[2] = 19.76, p < 0.0001).
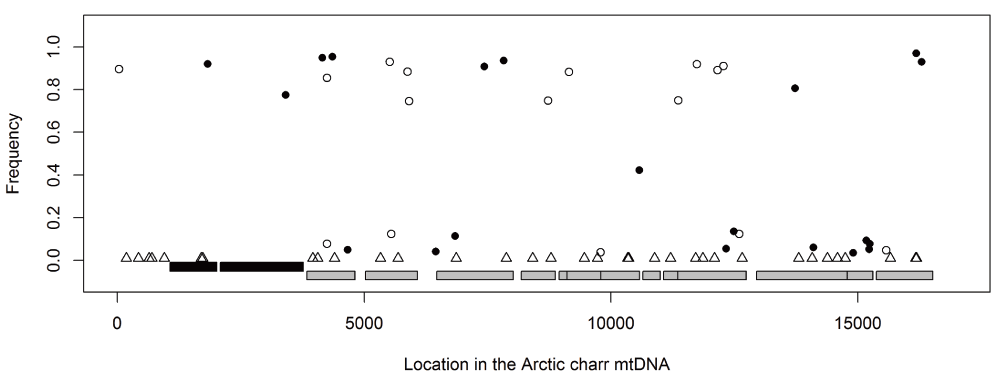
The frequency differences between morphs of candidate SNPs, estimated from the RNA-sequencing, graphed along the mtDNA chromosome. The SNPs indicate whether the derived allele is of higher frequency in SB (black dots) or AC (open circles). Sites of divergence between the Icelandic stocks and the Canadian reference sequence are indicated by triangles. The two black boxes represent the 12s (left) and 16s (right) rRNA genes, and gray boxes the 14 coding sequences.
In order to gauge the potential functionality of those variants we aligned the rRNA genes from nearly hundred fishes and several vertebrates. The position affected by m1829G>A and m3211T>C, in the 12s and 16s rRNAs, are not well conserved in fishes or vertebrates (Figure 7B and Figure 7D). However m3411C>T, in the 16s rRNA, alters a position that is nearly invariant in 100 fish genomes (Figure 7F). The only exception is Pacific menhaden, which curiously also has T in this position. This region could not be aligned properly in other vertebrates. Thus m3411C>T alters a conserved position, but probably not very drastically as the introduced allele is tolerated in another fish species.
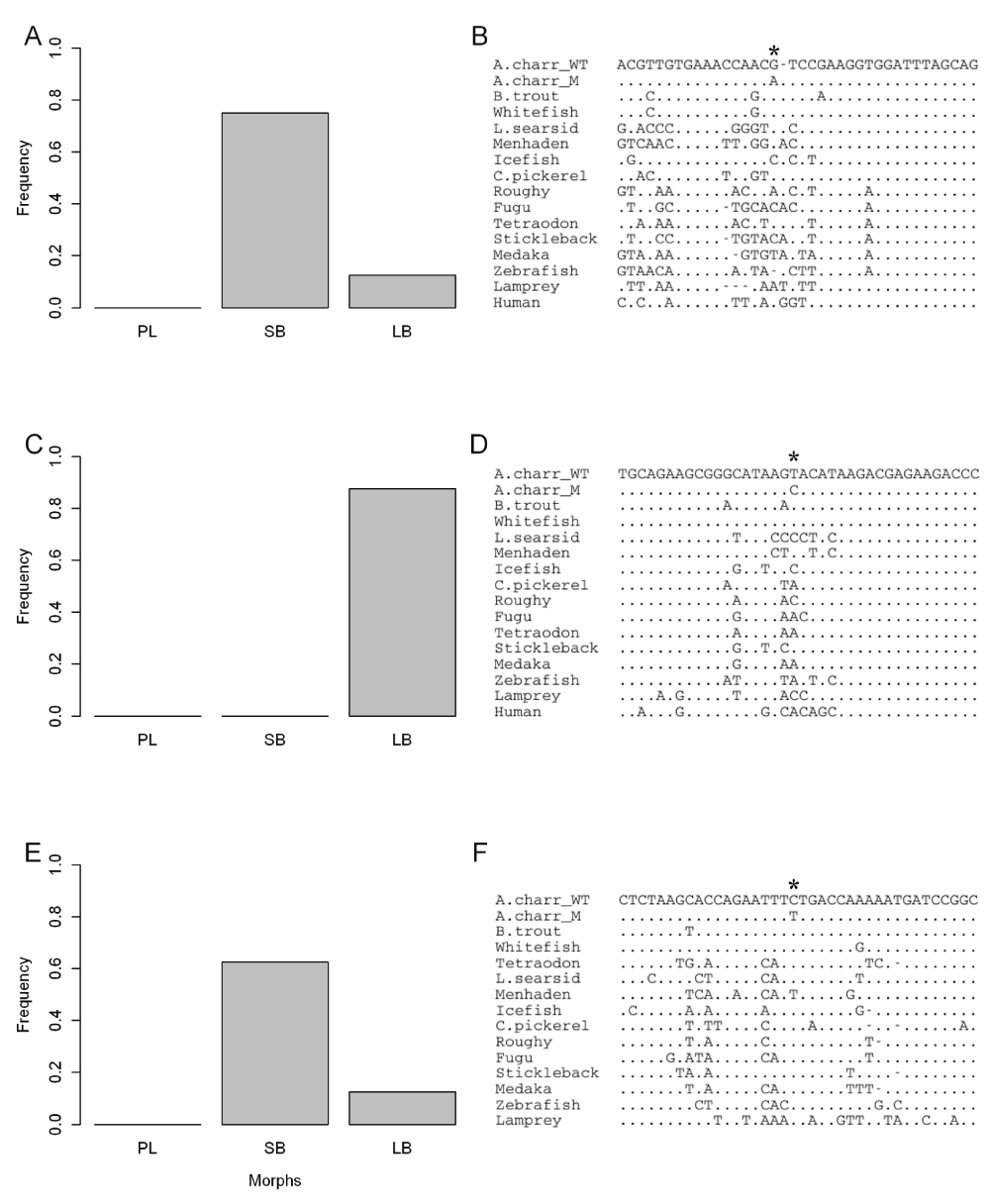
Aligned are several fish genomes, with Lamprey or humans as outgroups, reflecting a 38 bp window around each of the 3 positions (*). A, C, E) Frequency of each of those variants in three Arctic charr populations from Lake Thingvallavatn (PL, LB and SB). A total of 8 individuals were genotyped from each morph, see methods. B) Alignment of variant m1829G>A in the 12s rRNA gene in fishes, using humans as an outgroup. D) Similar alignment of a 16s variant, m3211T>C and F) alignment of variant m3411C>T in the 16s rRNA gene.
We are interested in how predictable evolution is a the molecular level and if there certain principles influence the rewiring of developmental and regulatory systems during evolution81. One way to study this is to identify genetic and developmental effects affecting key traits in species or populations which exhibit parallel evolution. The objective of this study were to get a handle on genetic and molecular systems that associate with benthic morphology in charr by mainly focusing on the small benthic morph in Lake Thingvallavatn, Iceland. To this end we performed transcriptome analysis contrasting the development of embryos from SB-charr and aquaculture charr.
As no reference genome is available for Arctic charr, we mapped reads to S. salar EST-contigs63 in order to estimate expression and identify candidate genetic polymorphisms. As many of the contigs are short or have overlapping annotations, we collapsed genes into paralogous genes when appropriate for the expression analysis. The main advantage of this approach was the reduction of the number of statistical tests (and hence an increase in statistical power). The downside is that paralog-specific expression patterns are masked, as our qPCR results of the natterin like gene family show (Figure 3 and S1 Figure). Recent rainbow trout data shows most paralogs from the latest whole genome duplication event retain the same expression pattern32 indicating that this scenario is probably uncommon; hence it is of considerable interest when two paralogs show distinct expression patterns82. In their analysis of the Arctic charr gill transcriptome, Norman et al. (2014)16 also used Illumina sequencing technology to evaluate expression. Their reads were longer (2x100 bp) than in this study (36 bp) enabling them to assemble contigs. They did not consider the paralogs in their approach and merged contigs based on sequence identity. Thus the complexity of Arctic charr transcriptome still remains a mystery that advances in sequencing technology, assembly algorithms and genome sequencing of this species could aid in revealing.
Our data reflects differential deployment of several gene classes during Arctic charr development. Studies in salmonids and other fish have demonstrated large changes in expression during early development, including coordinated changes in many cellular and developmental systems12,19,83–85. Several blood coagulation factors genes showed significant changes during charr development, and were also more highly expressed in the SB-charr. This might reflect differences in the rate of development of blood composition, or tissue composition, in the two morphs. Our main interest is on the derived and repeatedly evolved small benthic charr. For this study we chose AC-charr as a point of reference for several reasons, i) it has limnetic like head morphology, ii) availability, and iii) because we wanted a strong contrast in this first survey of charr developmental diversity. The AC-charr proved a useful, as the data presented here has already revealed differential expression of several developmental genes and regulators with differential expression between benthic and limnetic charr58,59. Furthermore we previously found tight correlation of RNA-seq expression and qPCR estimates - in this very same transcriptome58. Furthermore, we have actually used the same morphs (AC and SB) and samples in a comparison of the developmental miRNA transcriptome – which reveal that expression of several miRNAs correlates with morph differences62.
The genetic separation in two immunity genes among sympatric morphs in Lake Thingvallavatn52 prompted us to examine further the expression of Lyz and nattl that were differentially expressed between morphs.
Both genes are expected to be involved in immune defenses and had higher expression in SB. The substrate of lysozyme86 is the bacterial cell wall peptidogly-can and it acts directly on Gram-positive bacteria87. Lysozyme also promotes the degradation of the outer membrane and therefore indirectly acts also on Gram-negative bacteria88. Another gene that caught our attention was natterin-like. Natterins were first discovered from the venom gland of the tropical toxic fish species Thalassophryne nattereri75,76, and are found by sequence similarity in e.g. zebrafish, Atlantic salmon and here in Arctic charr. The predicted Natterin proteins contain a mannose-binding lectin-like domain (Jacalin-domain) and a pore-forming toxin-like domain and can cause edema and pain due to kininogenase activity75. Mannose-binding lectins are pathogen recognition proteins (antibodies) and therefore are important for the acute phase response of fish89,90. Our data suggest an immune related function of nattl genes in charr, as the highest expression was found in skin and kidney. This needs to be verified. It is possible that higher expression of those two genes in SB-charr reflect preparation of juveniles for bottom dwelling habitats, which may be rich in bacteria and challenging for immune systems.
In this study we collapsed contigs into gene or paralog groups for the transcriptome analyses. The disadvantage of this approach is that differential expression in one paralog, can be masked by other related genes that do not differ between groups or have contrasting expression patterns. We looked at this by studying the expression of three paralogs of the natterin like genes in different morphs during Arctic charr development, and among tissues of adult AC-charr. The data show that the three nattl genes are expressed differentially between the morphs, thus it is not divergence in the expression of one paralog that explains the general nattl expression disparity in the transcriptome. Certainly, other scenarios could apply to other genes in the transcriptome.
A study of the skulls of charr post-hatching embryos and juveniles from Lake Thingvallavatn, showed that some elements of the developing head ossified earlier in SB-charr than in PL-charr91. Our new data also demonstrate differences in craniofacial elements between AC- and SB-charr, along a limnetic vs. benthic axis79. Based on those differences between benthic and limnetic charr, we investigated further genes with roles in craniofacial development that were differentially expressed in the transcriptome. Guided by this transcriptome we had already found two extra-cellular matrix (ECM) remodeling genes, Mmp2 and Sparc and a conserved co-expression module of genes with known roles in craniofacial morphogenesis, to have higher expression in developing heads of benthic Arctic charr morphs than in limnetic morphs58,59. Bioinformatic and qPCR analyses suggest the co-expression module may potentially be affected by quantity of the transcription factor ETS2. These studies and the current data confirm the utility of the contrasting developmental transcriptomes for identifying candidate genes with differential expression during head development, as 7 out of 8 candidates were confirmed by qPCR. These genes had consistently higher expression in the developing head of two benthic morphs (SB and LB), and lower in more limnetic fish (AC and PL). This is striking, as three of the morphs (SB, LB and PL) studied are closely related and live in sympatry in Lake Thingvallavatn52.
We focused on a few targets of Tgf-β and Ahr signaling pathways because of their role in craniofacial morphogenesis and transcriptional connection92–94. Adseverin (Scin) was one of the top differentially expressed genes (Table 1) and has roles in rearrangements of the actin cytoskeleton, chondrocyte differentiation and skeletal formation95,96. Also, in the transcriptome Lsr, Cldn4 and Tgfbr2 had higher expression in SB-charr, and we show that higher expression of those genes associated with the benthic morphotype. Lsr is a molecular component of tri-cellular tight junctions97 and has been shown to be suppressed upon Tgf-β1 stimulation98 in a human cell line. Similarly, Cldn4, a tight junction protein with unknown role during embryonic morphogenesis, is a target of the Tgf-β and Ahr signaling pathways99,100. Finally, the expression of Tgfbr2, encoding a receptor of Tgf-β was slightly but significantly higher in the head of benthic morphs. Previous studies suggest a crucial role of Tgfbr2 in craniofacial morphogenesis101.
We also confirmed differential expression of other genes, including two with higher expression in SB-charr. Mvp is the predominant component of cytoplasmic ribonucleoprotein structures called vaults102, which is highly conserved across eukaryotes. The vaults have been something of an enigma, but are implicated in several processes from signal transmission and immune response103. The Jup gene also showed higher expression in SB-charr. Finally, higher expression of Vdra, encoding the vitamin D receptor A, was found in the heads of benthic forms. The receptor regulates mineral homeostasis, osteoblast differentiation and bone metabolism104.
To summarize, the results show that RNA-sequencing of Aquaculture charr with limnetic craniofacial morphology and small benthic charr can be used to reveal differential expression of genes that associate with limnetic and benthic divergence in craniofacial elements in sympatric charr morphs. It would be interesting if expression of these genes associates with benthic morphology in independently evolved charr populations, or even in other species with similar trophic diversity.
By comparing AC and SB-charr, that represents a small benthic resource morph that has evolved repeatedly in Icelandic stream and pond habitats51, we hoped to implicate genes and pathways involved in adaptation to these special habitats. The data point to differences between SB and AC-charr in systems related to energy metabolism, as may be expected considering their contrasting life histories. First, there is 2X higher expression of respiratory electron transport chain components in AC compared to SB-charr and 100% more mitochondrial derived reads are found in the AC-charr samples. Note that the direction of divergence is unknown, i.e. whether expression was up in AC or down in SB. Second, many derived candidate-SNPs in genes related to mitochondrial function were at high frequency on the AC branch. For instance in S100A1, which has been implicated in mitochondrial regulation in cardiac tissue in humans105, but its expression is probably not exclusive to this tissue. Third, while the mitochondrial ribosomal genes generally evolve slowly, we do see derived variants at high frequency in the SB and large benthic charr in Lake Thingvallavatn. Specifically, m3411C>T in SB affects a position that is highly conserved among fish, and could affect function of the 16s rRNA. Earlier studies of mitochondrial markers in S. alpinus did not find large signals of divergence within Iceland47,49,52, probably because they studied other genes. In summary, the results suggest divergence in mitochondrial function, due to the domestication of aquaculture charr and/or possibly reflecting adaptation of the small benthic charr in Lake Thingvallavatn.
The mitochondrion is more than a powerhouse, it integrates metabolism, cell cycle and apoptosis106. The number of mitochondria and its functions are known to correlate with environmental attributes. For instance in Antarctic fishes under extreme cold, higher numbers of mitochondria are found in muscle and heart cells107. Our data suggest an expression difference between morphs that could reflect differences in total number of mitochondrion, the number of mtDNA copies per mitochondrion or cell, or difference in RNA expression from the mtDNA, possibly due to evolution of mtDNA related to diet and/or temperature108. Further work is needed to map out the expression differences of mitochondrial related genes in more SB and anadromous charr morphs (representing the ancestral state). The mtDNA signals could also be investigated in populations along ecological clines (e.g. temperature) or with respect to life history109.
The data presented here set the stage for future investigations of the molecular and genetic systems involved in the development and divergence of the highly polymorphic and rapidly evolving Arctic charr. The results suggest genetic and expression changes in multiple systems relate to divergence among populations. The data reveal differential expression of two immunological genes between morphs and of several craniofacial developmental genes, that may help sculpture benthic vs. limnetic heads. The genetic data suggest among other things differentiation in the charr mtDNA between morphs. Our broad interest is in how natural selection tweaks genetic regulatory systems, for instance via genetic changes in regulatory sequences or post transcriptional modifiers relating to adaptations. Genetic changes affecting gene expression can be raw material for adaptation, but could also rise in frequency due to reverberations in regulatory cascades81. Our specific aim was to cast light on the developmental and population genetics of the unique small benthic charr, typically found in cold springs and small pond habitats in Iceland, particularly those with lava substratum36,51. The availability of charr populations at different stages of divergence sets the stage for future genomic studies of the roles of genes, environment and plasticity for shaping this polymorphic species.
F1000Research: Dataset 1. Parameters and multiple testing corrected p-values for expression analysis, 10.5256/f1000research.6402.d48005110
F1000Research: Dataset 2. qPCR data for tests of expression in charr developing embryos and adult tissues., 10.5256/f1000research.6402.d48006111
F1000Research: Dataset 3. qPCR data for tests of expression in charr developing embryo heads., 10.5256/f1000research.6402.d48007112
Conceived and designed the study: JG, AP, ZOJ, SSS, SRF, VHM, EPA.
Sampling, crosses and rearing: SSS, BKK, ZOJ, KHK, VHM, AP.
RNA extraction and RNA sequencing: SRF.
Analyses of RNA sequencing data: JG, AP.
qPCR work: EPA, SSS2, VHM.
SNP analyses: JG, AP.
SNP confirmation: IMJ, KHK, AP.
Comparative genomic analysis: AP.
Writing: AP, JG, EPA, VHM, SSS.
Analyses: JG, AP, EPA, SSS2.
Gathered the data: ZOJ, SRF, EPA, IAJ, KHK, SSS2.
This project was supported by The Icelandic Center for Research (grant number: 100204011) to SSS, AP, ZOJ and BKK, The University of Iceland Research/Doctoral Fund to JG and KHK and University of Iceland research fund to AP, SSS and ZOJ.
I confirm that the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We would like to thank Baldur Kristjansson for help with genomic alignments. We are very grateful to Droplaug N. Magnusdottir, Gudbjorg Th. Orlygsdottir, Steinunn Snorradottir and Olafur Th. Magnusson at deCODE Genetics for help with the Illumina sequencing.
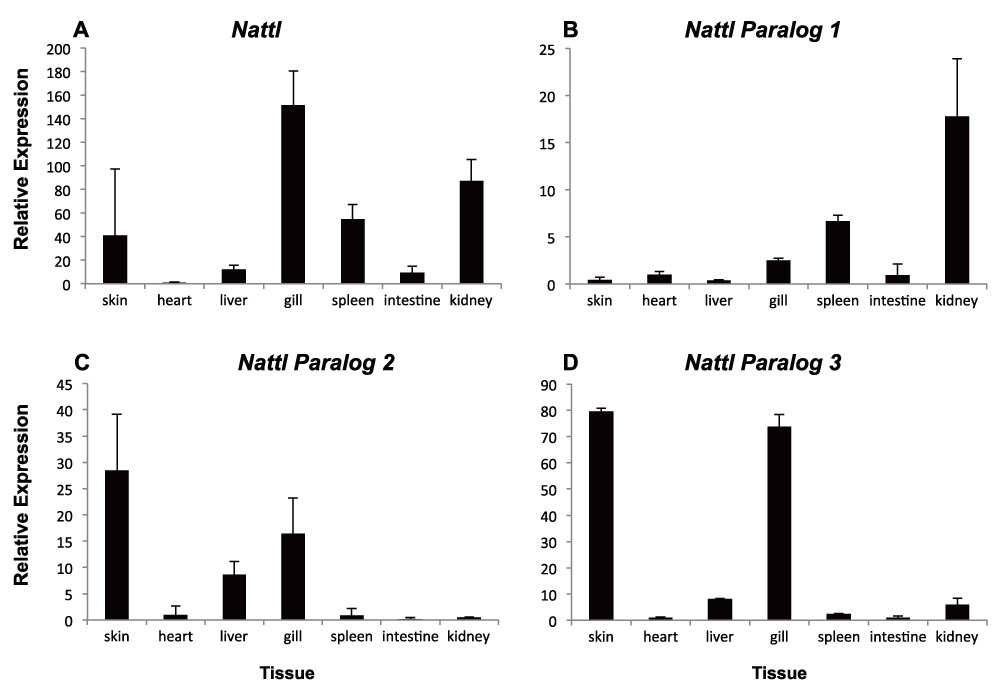
Relative expression of Natterin (A) & Natterin paralogs 1–3 (B–D) within different tissues (skin, heart, liver, gill, spleen, intestine & kidney) of adult aquaculture charr (RT-qPCR); expression plotted for different tissues, relative to heart tissue (lowest expression levels).
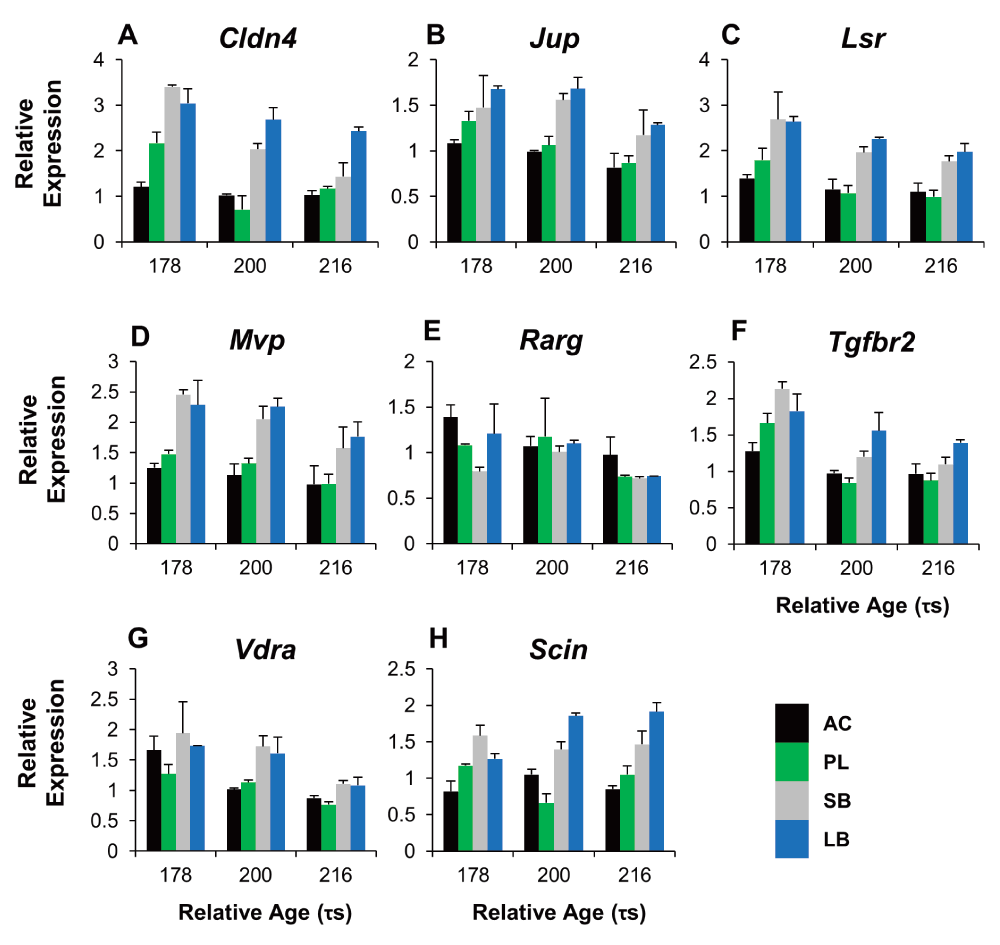
Relative expression of 12 candidate genes with characterized craniofacial expression during zebrafish development (ZFIN website) in the head of SB, LB, PL and AC at three time points in development. In the transcriptome data all of the genes had shown higher expression in SB at 200 τs. The expression is normalized to the geometric means of two craniofacial reference genes (ACTB and IF5A1). Expression is relative to a replicate of AC morph at 200 (τs), set to one. Error bars represent standard deviation calculated from two biological replicates and each biological replicate contains homogenate of six heads.
Primer sequences, melting temperatures and primary data.
Numbers of reads aligning to salmon reference for each sample.
Expression of nine genes was analyzed in whole SB and AC-charr embryos, at two developmental timepoints (161 and 200 day degrees).
ANOVA for relative expression levels of Natterin-like and Natterin-like paralogs 1–3 in Arctic charr whole embryos (among SB, AC and PL morphs) and tissues from adult AC-charr.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)