Keywords
Alzheimer’s, Apolipoprotein E, Aβ, liver X receptor, retinoid X receptor, Bexarotene
This article is included in the Preclinical Reproducibility and Robustness gateway.
Alzheimer’s, Apolipoprotein E, Aβ, liver X receptor, retinoid X receptor, Bexarotene
Alzheimer’s disease (AD) is a debilitating neurodegenerative disease and the leading cause of dementia in the elderly. It is currently estimated that 5 million people in the US and 30 million worldwide are afflicted with this disease. The pathological hallmarks of AD are the presence of extracellular amyloid plaques and intracellular neurofibrillary tangles in the hippocampus and cortical areas of the brain1. The core constituent of the amyloid plaques is a 4 kDa peptide known as amyloid-β peptide (Aβ). Aggregation of Aβ into soluble, multimeric assemblies and insoluble amyloid fibrils is hypothesized to contribute directly to the pathogenesis of AD; therefore therapeutic strategies aimed at lowering soluble Aβ levels in the brain would be predicted to have a disease-modifying effect2.
The E4 allele of apolipoprotein E (APOE) is the largest genetic risk factor for sporadic, late-onset AD. The presence of a single copy of E4 increases the risk for Alzheimer’s disease 3-fold and individuals with 2 copies are 15 times more likely to develop AD3. Data showing that APOE4 carriers begin to accumulate amyloid deposits earlier in life relative to non-carriers4 has led to the hypothesis that increased risk associated with an E4 genotype may be the result of the effects of APOE on Aβ production, turnover and/or clearance from the central nervous system (CNS).
The expression of genes encoding lipid-transport proteins, including APOE is transcriptionally regulated by the ligand-activated nuclear receptors, peroxisome proliferator-activated receptor gamma (PPARγ) and liver X receptors (LXRs) which form obligate heterodimers with retinoid X receptors (RXRs)5. Additionally, activation of these receptors has been shown to affect the activation state of macrophage and microglia6. Based on the processes influenced by this nuclear receptor family it is a reasonable hypothesis that agonism of one or more members of the family could have beneficial effects on Aβ homeostasis in the CNS. In fact, several groups have demonstrated that agonism of the LXR receptor resulted in reduced amyloid plaque burden and/or soluble Aβ levels in amyloid precursor protein (APP) transgenic mouse models7–9. More recently it was reported that a highly selective, blood-brain barrier–permeant, RXR agonist, Bexarotene (Targretin), resulted in enhanced clearance of soluble Aβ in an APP transgenic mouse model in an APOE-dependent manner. In addition, Aβ plaque burden was reduced more than 50% within 72 hours. Further, Bexarotene treatment also resulted in a similar reduction (~25%) in brain interstitial fluid (ISF) levels of Aβ in non-transgenic, C57Bl/6 mice 7–12 hours following a single administration10.
In the following study, we sought to examine the effects of LXR/RXR agonism on Aβ homeostasis in the CNS of non-transgenic rats using the RXR agonist, Bexarotene and the LXR agonist, TO901317.
In vivo pharmacodynamic studies: All procedures were approved by the Amgen Institutional Animal Care and Use Committee. Young male Sprague-Dawley rats (175–200 g) were purchased from Harlan (Indianapolis, IN) and were maintained on a 12h light/dark cycle with unrestricted access to food and water until use. Rats were dosed orally for 3 and 7 consecutive days with AMG8155, a proprietary small molecule BACE1 inhibitor, at 3 mg/kg in 2% HPMC and 1% Tween 80, pH 2, Bexarotene (Alfa Aesar, Ward Hill, MA) at 100 mg/kg in 30% Labrasol, 1% Tween 20, 2% Providone and 0.05% BHA, pH7.0 (Vehicle 3), and TO901317, a LXR agonist (Fisher Scientific, Pittsburgh, PA), at 30 mg/kg in 0.5% NaCl, 2% Tween 80 (Vehicle 4). 4 hours post dose on the last day of study, rats were euthanized with CO2 inhalation for 2 minutes and the cisterna magna was quickly exposed by removing the skin and muscle above it. Cerebrospinal fluid (CSF) was collected with a 30 gauge needle inserted through the dura membrane covering the cisterna magna. CSF samples with visible blood contamination were discarded. Blood was withdrawn by cardiac puncture and plasma was obtained by centrifugation at 15,000 rpm for 10 min at 4°C for drug exposure. Brains were removed and, along with the CSF, immediately frozen on dry ice and stored at -80°C until use. The frozen brains were subsequently homogenized in 10 volumes (w/v) of 0.5% Triton X-100 in TBS with protease inhibitors cocktails. The homogenates were centrifuged at 355,000 rpm for 30 min at 4°C.
Quantification of Aβ40 and APOE in brain and CSF: Samples are analyzed for Aβ levels by immunoassay with a MSD imager. Briefly, 96-well avidin plates (MesoScale Discovery, Inc., Gaithersburg, MD) were coated with biotinylated-anti-Aβ antibody 4G8 (mouse monoclonal, Cat# Sig 39240-1000, Covance Research Products, Princeton, NJ) at 10 μg/ml in PBS. Samples were co-incubated in the plate overnight at 4°C along with a ruthenium-labeled anti-Aβ antibody specific for the C-terminal region of Aβ40 (ConFab40; Amgen, Thousand Oaks, CA). Plates were then washed, 150 μl/well read buffer T (MesoScale Discovery, Inc.) was added, and plates were read immediately on a Sector 6000 imager according to the manufacturer’s recommended protocol (MesoScale Discovery, Inc.). All samples were assayed in triplicate and analyzed by using Prism version 5.04 (GraphPad Software Inc., San Diego, CA). Data was analyzed by one-way analysis of variance and Dunnett’s multiple comparison test.
APOE levels in brain (50 μg homogenates) and CSF (10 μl) were analyzed by Western blot following PAGE using 4–12% Bis-Tris gels (Invitrogen, Carlsbad, CA). Blots were probed with primary antibodies to APOE (goat polyclonal, EMD Millipore; 1:1000) and the loading control, actin (ThermoFisher Scientific; 1:200) for 60 min at 4°C and then washed with TBST (Tris-buffered saline, 0.1% Tween 20) three times at room temperature, followed by (Goat-anti-mouse) secondary antibody (ThermoFisher Scientific; 1:1000) for 30 min at 4°C. Densitometric analysis of ApoE was performed (exposure time of 4 minutes with a relative intensity of 2.0, Odyssey imaging system, with application software Version 3.0) followed by an unpaired t-test using GraphPad Prism 5.04 software.
Measurement of Plasma, CSF, and Brain Drug Concentration: Aliquots of plasma (50 μl) were combined with 300 μl of acetonitrile containing 125 μl structurally related internal standard (IS), vortexed, and centrifuged. Supernatant was transferred into a plain polypropylene 96-well plate for sample analysis. Brain tissue samples were homogenized by using a Covaris (Woburn, MA) acoustic homogenizer. Aliquots of 50 μl homogenate were combined with acetonitrile containing a structurally related IS, vortexed, and centrifuged at 1,900 g for 5 minutes. Supernatant was transferred into a 96-well plate for sample analysis. Analytical standards and tissues were measured by liquid chromatography mass spectrometry (Shimadzu Pumps Autosampler Prominence for HPLC and PE Sciex API 4000 for MS, with Analyst 1.6.1 software) using atmospheric-pressure chemical ionization and multiple reaction monitoring in the positive ion mode.
Our aim in this study was to investigate the effects of RXR/LXR agonism on Aβ homeostasis in the CNS of non-transgenic rats using the RXR agonist, Bexarotene and the LXR agonist, TO901317. As a positive control, we included a β-secretase inhibitor (AMG8155). Compounds and appropriate vehicle controls were administered to naïve Sprague Dawley rats at doses indicated in Table 1 for either 3 or 7 consecutive days.
| Group | Dose (mg/kg) |
|---|---|
| Bexarotene | 100 |
| AMG8155 | 3 |
| TO901317 | 30 |
Table 1 lists the 3 compounds tested in this study along with the respective doses (mg/kg).
Following 3 and 7 days of dosing, animals were evaluated for both compound levels and pharmacodynamic endpoints. APOE levels were quantitated in brain homogenate and CSF by Western blot. Aβ40 levels were quantitated in the same compartments using immunoassay as described in the Materials and methods section. Following 3 and 7 days of dosing, APOE levels were increased in brain and CSF in the TO901317 treated animals compared to vehicle treated animals (Figure 1). Changes in CSF were statistically significant at both 3 (p = 0.0002) and 7 days (p = 0,0007) whereas changes in brain were statistically significant at day 3 (p = 0.030) but did not reach significance at day 7 (p = 0.056). Bexarotene treatment also resulted in a statistically significant increase in CSF APOE levels compared to vehicle treated animals following both 3 (p = 0.019) and 7 days (p = 0.002) of dosing (Figure 2). APOE levels in brain following Bexarotene treatment trended towards an increase however these changes were not statistically significant. Soluble Aβ40 levels were unchanged in brain and CSF following 3-day (Figure 3) and 7-day (Figure 4) treatment with either Bexarotene or TO901317. The positive control BACE inhibitor, AMG8155 effectively reduced Aβ40 levels by 70% and 71% in CSF and by 67% and 69% in brain in the 3-day and 7-day studies respectively (Figure 3 and Figure 4).
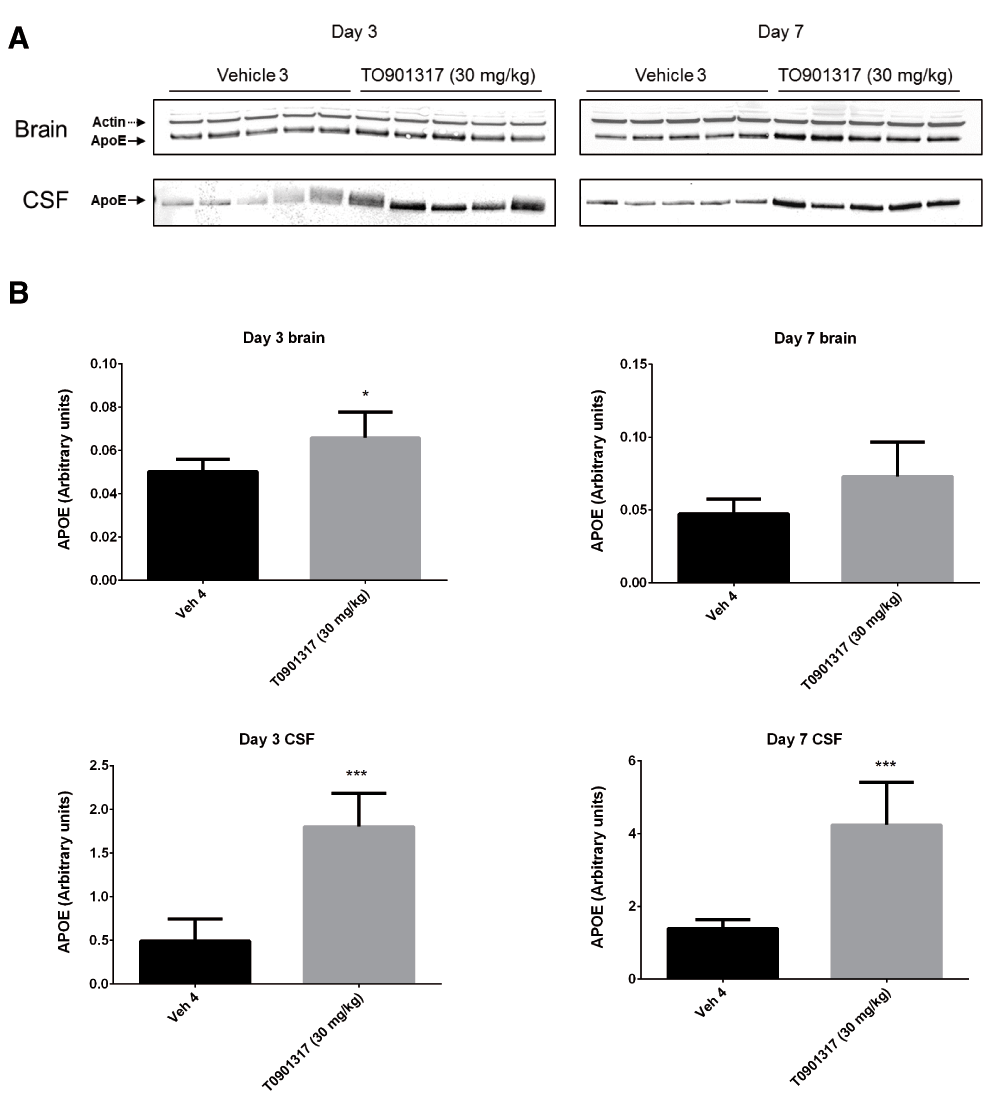
APOE was also increased in brain however the changes only reached statistical significance at day 3. A) Western blot analysis of APOE in brain and CSF. B) Densitometric analysis of the bands was performed as described in the Materials and Methods section; data are presented as the mean plus standard deviation; Vehicle 4 (black bars) and TO901317 (gray bars).
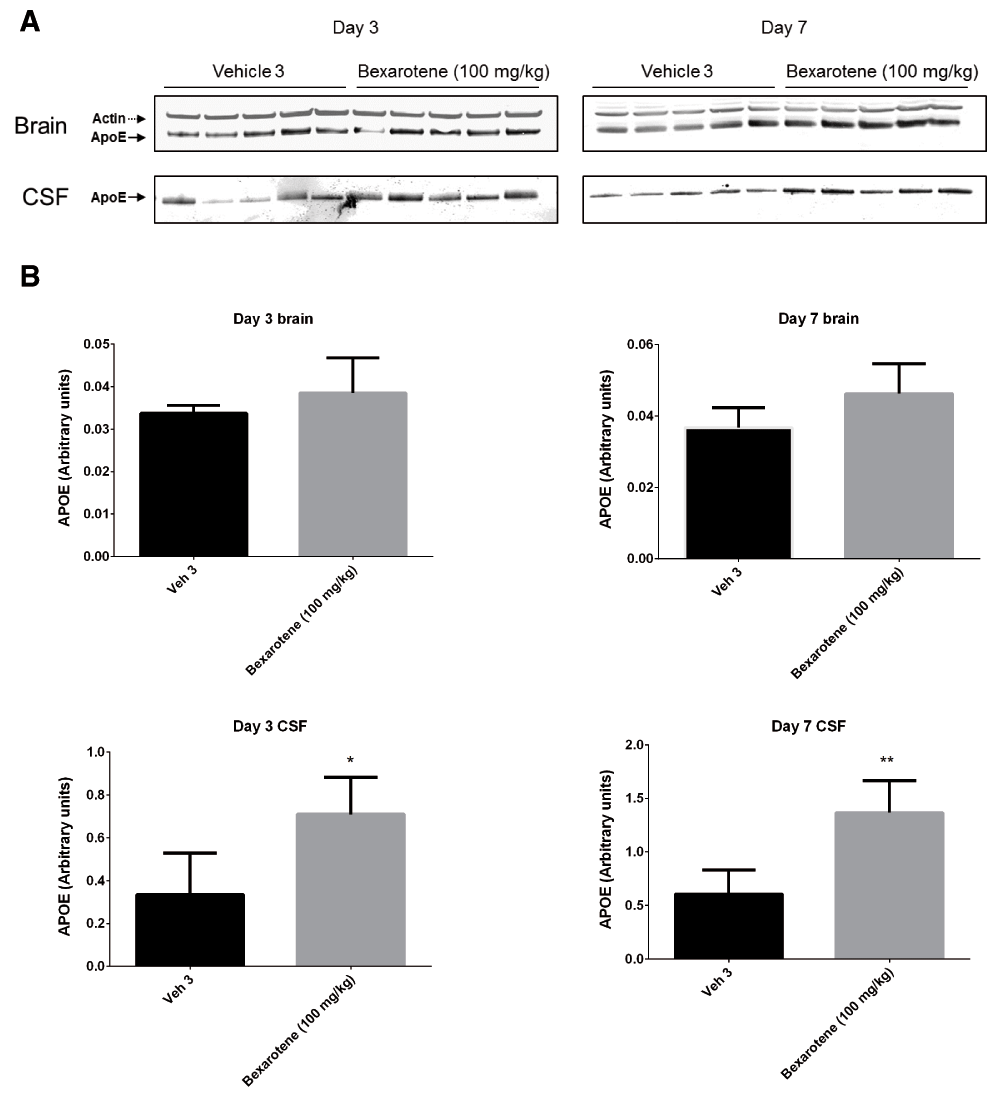
APOE changes in brain were not statistically significant. A) Western blot analysis of APOE in brain and CSF. B) Densitometric analysis of the bands was performed as described in the Materials and Methods section; data are presented as the mean plus standard deviation; Vehicle 4 (black bars) and Bexarotene (gray bars).
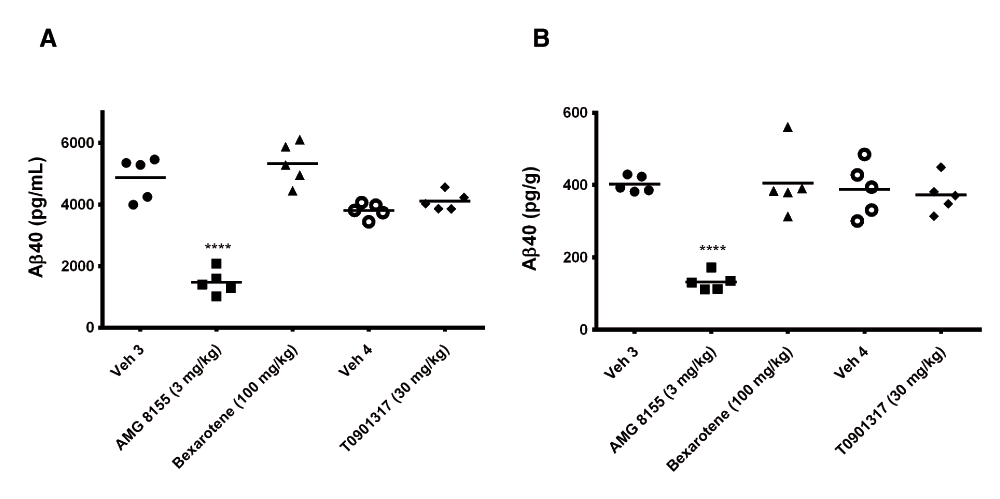
Aβ40 levels in (A) CSF and (B) brain were unchanged following 3 days of treatment with Bexarotene (triangles) or TO901317 (diamonds). Positive control BACE inhibitor AMG8155 (squares) reduced Aβ40 levels 70 and 67% in CSF and brain respectively following a single administration.
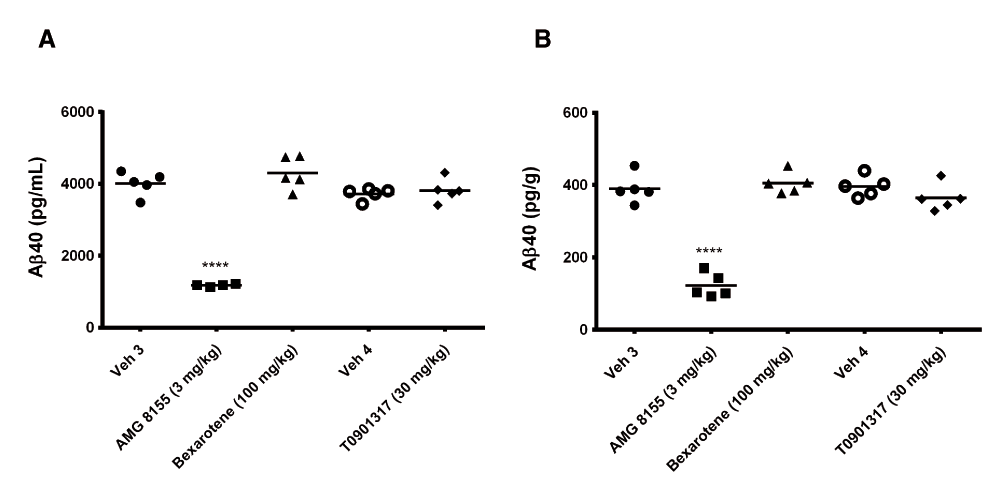
Aβ40 levels in (A) CSF and (B) brain were unchanged following 7 days of treatment with Bexarotene (triangles) or TO901317 (diamonds). Positive control BACE inhibitor AMG8155 (squares) reduced Aβ40 levels 71 and 69% in CSF and brain respectively following a single administration.
Drug levels of Bexarotene and TO901317 were measured in plasma and brain homogenate following 3 and 7 days of dosing (Table 2). Total levels of both compounds achieved single-digit to low double-digit μM levels in brain and showed good uptake in brain relative to plasma in both dosing paradigms.
In this study we demonstrate that 3-day or 7-day treatment of naïve rats with the LXR agonist, TO901317 or RXR agonist, Bexarotene treatment resulted in an increase in APOE levels in CSF. No changes were observed in CSF or brain Aβ40 levels with either compound after 3 or 7 days of dosing. We hope that these findings will stimulate further discussion in the Alzheimer’s research community on the impact of LXR/RXR agonism on central Aβ homeostasis.
Open Science Framework: Dataset: Effect of LXR/RXR agonism on brain and CSF Aβ40 levels in rats, doi: 10.17605/OSF.IO/3NS6411
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
The formulation issue was explicitly discussed in the literature and the FDA filing for bexarotene and was detailed on our ‘Response’ to the ‘ Comments’ in Science (Landreth et al., 2013). The Amgen scientists (and others) clearly did not make an effort to understand and replicate the original study design, nor appreciate the importance of the formulation of the drug as it relates to nuclear receptor action. Although the Amgen study was narrowly focused on ApoE and soluble Abeta 40 and did not examine other aspects of our work, the same considerations apply to other contested outcomes of our study.
The Amgen study employed wild type Sprague-Dawley rats, whereas all other studies with bexarotene used murine models of AD. A study of the LXR agonist TO9001317 in rats that was similar to that of Amgen was published by Merck (Suon et al., 2010) and was only partially replicated in the Amgen study and was not cited.
Our work on bexarotene has led to clinical examination of its effects in Alzheimer’s disease. Cummings and colleagues published the outcome of a phase II study of bexarotene in mild to moderate AD patients last week (Cummings et al., 2016). While bexarotene treatment did not have a demonstrable effect in individuals possessing an ApoE4 allele, individuals that did not have an ApoE4 gene (representing about 80% of the population) exhibited a robust reduction in brain amyloid with a parallel appearance of Abeta42 in plasma. Thus, a brief 30 day treatment altered two canonical biomarkers of AD in patients with diagnosed disease. These findings are consonant with a recent case report reporting cognitive improvement with bexarotene treatment (Pierrot et al. 2015). I hasten to add that the small study size precludes making any hard conclusions from this trial and they should be interpreted cautiously. However, these preliminary clinical findings support and validate our original report using mouse models of the disease.
I don't think the Amgen study adds anything to what was previously known. The work has a logical flaw that undermines the conclusion that they were unable to repeat our study outcomes, including the use of a different model. I think this study is emblematic of the problems associated with reporting ‘failure to replicate’ findings in studies that do not genuinely reproduce the published work.
The formulation issue was explicitly discussed in the literature and the FDA filing for bexarotene and was detailed on our ‘Response’ to the ‘ Comments’ in Science (Landreth et al., 2013). The Amgen scientists (and others) clearly did not make an effort to understand and replicate the original study design, nor appreciate the importance of the formulation of the drug as it relates to nuclear receptor action. Although the Amgen study was narrowly focused on ApoE and soluble Abeta 40 and did not examine other aspects of our work, the same considerations apply to other contested outcomes of our study.
The Amgen study employed wild type Sprague-Dawley rats, whereas all other studies with bexarotene used murine models of AD. A study of the LXR agonist TO9001317 in rats that was similar to that of Amgen was published by Merck (Suon et al., 2010) and was only partially replicated in the Amgen study and was not cited.
Our work on bexarotene has led to clinical examination of its effects in Alzheimer’s disease. Cummings and colleagues published the outcome of a phase II study of bexarotene in mild to moderate AD patients last week (Cummings et al., 2016). While bexarotene treatment did not have a demonstrable effect in individuals possessing an ApoE4 allele, individuals that did not have an ApoE4 gene (representing about 80% of the population) exhibited a robust reduction in brain amyloid with a parallel appearance of Abeta42 in plasma. Thus, a brief 30 day treatment altered two canonical biomarkers of AD in patients with diagnosed disease. These findings are consonant with a recent case report reporting cognitive improvement with bexarotene treatment (Pierrot et al. 2015). I hasten to add that the small study size precludes making any hard conclusions from this trial and they should be interpreted cautiously. However, these preliminary clinical findings support and validate our original report using mouse models of the disease.
I don't think the Amgen study adds anything to what was previously known. The work has a logical flaw that undermines the conclusion that they were unable to repeat our study outcomes, including the use of a different model. I think this study is emblematic of the problems associated with reporting ‘failure to replicate’ findings in studies that do not genuinely reproduce the published work.