Keywords
IPCS (Inositol phosphorylceramide synthase), Sphingolipid metabolism, Leishmania, Leishmaniasis, Drug Inhibitor design, Coumarin derivatives, Molecular docking, Molecular dynamics simulation
This article is included in the Cheminformatics gateway.
This article is included in the Neglected Tropical Diseases collection.
IPCS (Inositol phosphorylceramide synthase), Sphingolipid metabolism, Leishmania, Leishmaniasis, Drug Inhibitor design, Coumarin derivatives, Molecular docking, Molecular dynamics simulation
IPCS – Inositol phosphorylceramide synthase, IPC – Inositol phosphorylceramide, AUR1 – Aureobasidin 1, DAG – Diacylglycerol, RMSD – Root Mean Square Deviation, LINCS – Linear constraint solver, PME – Particle Mesh Ewald.
Infectious disease, leishmaniasis, is the major cause of parasitic diseases affecting 12 million people worldwide. Most of the anti-leishmanial compounds do not have well-defined mechanisms. The first line treatment of cutaneous leishmaniasis involves the administration of antimony based compounds. Treatment of L. major amastigotes with Sb(V) has been found to induce apoptosis by the induction of oxidative-stress and increase in intracellular calcium1. Non-antimony based treatments such as miltefosine, and topical formulations of paromomycin are cost effective, convenient and less toxic than antimony based compounds. Amphotericin B being a liposomal formulation is expensive, has a low therapeutic index and is difficult to administer2. Newer formulations for the treatment of this disease include the administration of miltefosine. Miltefosine (hexadecylphosphocholine), originally an anticancer drug has been reported to induce apoptosis of L. major amastigotes in the infected macrophages3. Development of newer treatment modalities arise from the problem of drug resistance and quick adaptability of the parasite to the host immune response4–6.
Sphingolipids like IPC, form an important component of the parasitic membranes7. IPCS (inositol phosphorylceramide synthase) is an enzyme involved in the sphingolipid metabolism of protozoans and other fungal species8. The relative importance of IPCS in Leishmania has been identified through biochemical network modeling9. IPCS catalyzes the conversion of ceramide to IPC which forms the most predominant sphingolipid of the parasite10 (Figure 1). IPCS also maintains the concentration of DAG and ceramide, both of which serve as secondary messengers in several signal transduction events11. IPCS localizes into the lipid rafts of the Golgi complex12. Lipid rafts have been proposed to involve in a wide array of events like trafficking of lipid modified proteins in addition to playing an important role in the formation of signal transduction complexes13. IPCS has been important for maintaining the viability and the infectivity of several fungal species like Cryptococcus neoformans, Candida albicans and pathogens like Leishmania14–17. Interestingly there is no mammalian equivalent of this enzyme and the major sphingolipid in the host is sphingomyelin instead of IPC. Hence IPCS has been considered as a choke point enzyme in the sphingolipid metabolism of Leishmania thereby serving as a druggable target for the treatment of several fungal and protozoan diseases like leishmaniasis. LmjIPCS comprises of 338 amino acids and has 6 transmembrane domains and belongs to the PAP2c family9. IPCS is encoded by the AUR1 gene. IPCS protein present in fungi exhibits sensitivity to antifungal agents like galbonolide A, aureobasidin A, macrolidegalbonolide and khafrefungin18,19. IPCS has been recently discovered in Leishmania and to the best of our knowledge there are no reports of inhibitor design against this protein. This paper explores the possibility of targeting IPCS for the development of anti-protozoan compounds. An in silico approach for drug design has led to the development of five novel coumarin derivatives. The refinement and validation of the docked complexes has been done using molecular dynamics simulations to map the protein ligand interactions. Based on the in silico findings, the promising candidates were considered for further experimental evaluation and validation.
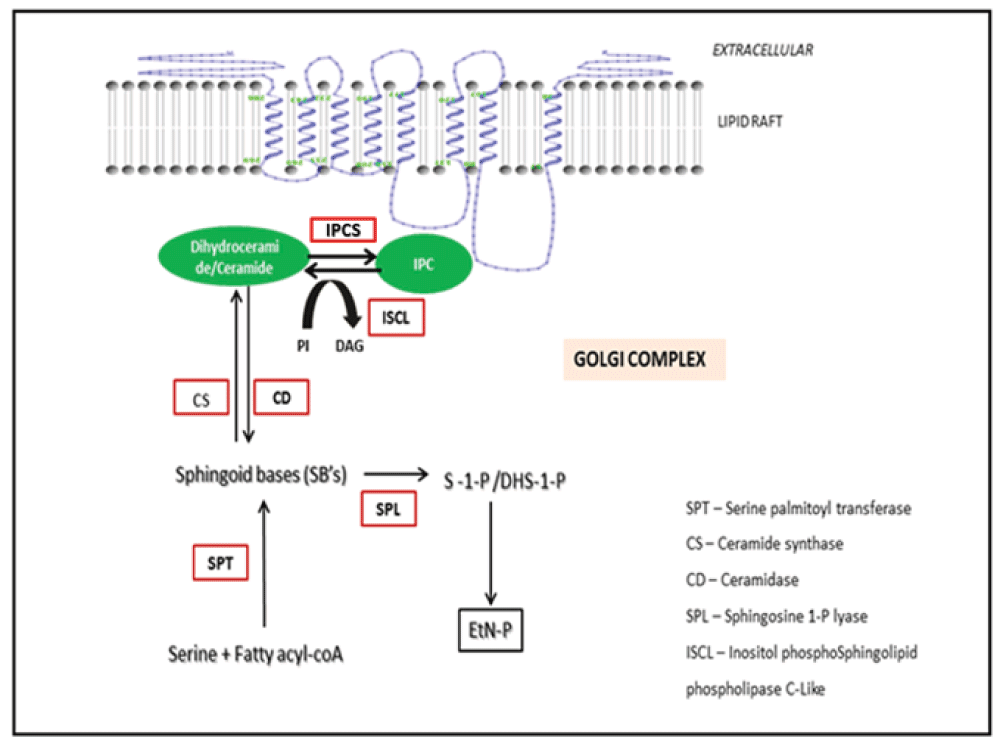
IPCS catalyzes the reaction involving the conversion of ceramide to IPC (Inositolphosphorylceramide). IPC forms the most predominant sphingolipid in Leishmania. IPCS plays an important role in maintaining the viability of the parasite.
A set of coumarin derivatives were prepared by the assembly of pharmacophoric groups. The 2D structures of the inhibitors were drawn and edited using Chemsketch version 12.0120 (Figure 2). The SMILES format for all the compounds was generated using Open Babel version 2.3.121. Inhibitors were designed and filtered using the “Lipinski rules of five”22 and Veber’s rules23 using the Molinspiration Property Calculation Service (www.molinspiration.com).
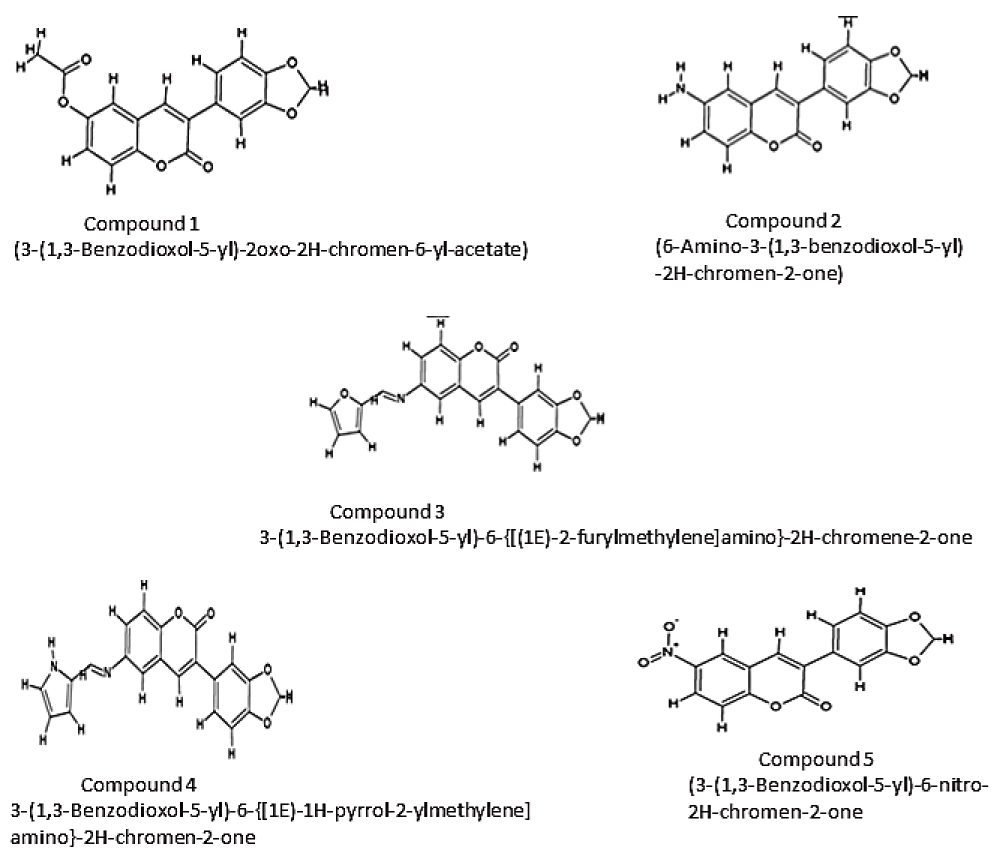
The designed inhibitors are Coumarin derivatives. Coumarin increases the phagocytic activity of the macrophages.
The pharmacophore models describing the inherent chemical features of the inhibitors were generated using the “Feature mapping protocol” available in Discovery Studio version 3.0. (www.accelyrs.com). Pharmacophore models of the inhibitors indicated that the ligand had at least a maximum of 5 pharmacophoric features i.e. Hydrogen bond acceptors (HBA), Hydrogen bond donors (HBD), positive ionizable groups (PI), Ring aromatic groups (RA) and the Hydrophobic groups (HY) present in the ligand.
IPCS is one of the emerging drug targets for the treatment of leishmaniasis. The crystal structure of the IPCS protein has not been solved and hence the 3D structure for the IPCS protein developed by our group before has been used for the inhibitor design. The model was developed using the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). The predicted model has a total of 338 amino acid residues and has 7 transmembrane helices9. The 3D structure of IPCS was energy minimized by the steepest gradient method of energy minimization using the GROMACS 4.0 package24. Mol2 file format of the inhibitors was converted to PDBQT format using MGL tools prior to docking. All the water and solvent atoms of the protein were removed prior to docking and the polar hydrogens were added. The protein was kept rigid while the ligand was allowed to rotate and explore more flexible binding pockets. Docking of the inhibitors onto the IPCS protein was performed using Auto Dock 4 version 1.5.6 and Auto Dock vina. version 1.1.2. The grid box size dimensions were 40X40X40, the default scoring function was used for docking25,26. Binding modes of the docked complexes were obtained and the amino acid residues present at a distance of 5Å were considered as the binding partners of the ligands. The interaction diagrams representing the docked complexes have been generated using Pymol v 1.3.
Molecular dynamics simulation is a computational method that provides information regarding the time dependent behavior of any molecular system by integrating Newton’s laws of motion. The docked complexes (IPCS-inhibitor complex) were subjected to MD simulation using Desmond version 4.4 (Schodinger Biosuite). MD simulation of both the IPCS protein and IPCS –ligand complexes were performed for a time period of 10ns by using the OPLS force field. The complex was centered in a cubic box and filled with TIP3P water molecules. The system was neutralized and the initial energy minimization for the system was done using the conjugant gradient algorithm. The Martyna-Tobias-Klein scheme was used for pressure coupling. Electrostatic forces were calculated using the PME algorithm27. All runs were performed at 300K at constant volume and temperature (NPT ensemble) under certain periodic boundary conditions. RMSD plots for the backbone atoms for both the protein and ligand bound protein were generated to understand the relative stability of the ligand inside its binding pocket and the IPCS-inhibitor complexes were visualized.
Macrophage cell population was collected post 24 h treatment with the compound 3, washed and suspended in 1XPBS. Cells were stained with 10µl of 10μg/mL of propidium iodide (PI) dye (Invitrogen) and acquired on FACS. Total macrophage population was gated based on their forward scatter (FSC) and side scatter (SSC) after excluding the cell debris. A minimum of 10,000 events were acquired for each sample on FACS Canto II (Beckon Dickson, San Jose, California) and analyzed using FACS Diva Software (version 6.2.1) (Beckon Dikson, San Jose, California).
A group of coumarin derivatives were prepared as inhibitors of the IPCS protein belonging to L. major. Assessment of the drug like properties indicated that all the inhibitors were found to comply with the Lipinski’s “Rule of five” (molecular weight (Mwt) ≤ 500, clogP ≤ 5, H-bond donors (HBD) ≤ 5, and acceptors (HBA) ≤ 10) and Verber’s rules (no. of rotatable bonds < 10, PSA ≤ 140A2) (Table 1).
HBA – Hydrogen bond acceptor, HBD – Hydrogen bond donor, HY – Hydrophobic, RA – Ring aromatic, MR – Molar refractivity, NROTB – No. of rotatable bonds, cLogP – log octanol/water partition coefficient, PSA – Polar surface area, NSC – No. of stereo centers.
| S.No | Mwt | cLogP | HBA | HBD | HY | RA | MR | NROTB | PSA(A2) | NSC |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 324 | 2.8 | 8 | 0 | 2 | 4 | 83.82 | 3 | 74.98 | 0 |
| 2 | 281 | 2.46 | 5 | 2 | 2 | 4 | 76.92 | 1 | 74.70 | 0 |
| 3 | 359 | 4.22 | 7 | 0 | 3 | 6 | 78.47 | 4 | 73.84 | 1 |
| 4 | 358 | 3.86 | 6 | 1 | 3 | 6 | 100.70 | 1 | 77.36 | 1 |
| 5 | 311 | 3.15 | 9 | 0 | 2 | 4 | 78.65 | 2 | 94.50 | 0 |
Molecular docking studies reveal the binding modes of the ligand with the IPCS protein giving an insight into the crucial amino acid residues that are involved during the binding. A comparison of the binding energies of all the compounds indicates that compound 3 has the least binding energy among all and hence exhibits maximum affinity towards the IPCS protein (Table 2). The interaction modes of all the IPCS inhibitors post docking along with their pharmacophoric features have been presented [Figure 3]. Binding mode analysis reveals that hydrophilic amino acids like Arg299 and His220 were found to be involved in hydrogen or π bonding with most of the ligands (Table 3). The relative stability of the compounds within the binding site was maintained due to the van der Waal’s interaction between the hydrophobic amino acids of the IPCS protein and the ligand (Table 4).
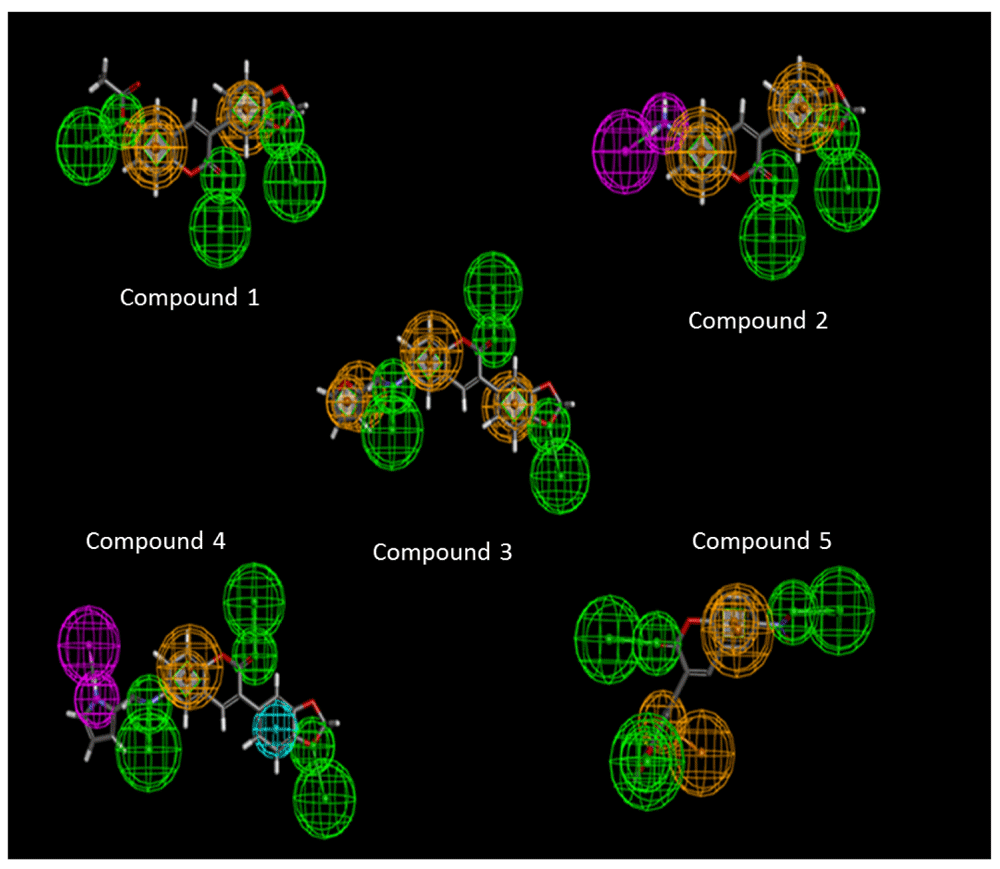
The pharmacophoric features such as hydrogen bond acceptors (green), hydrogen bond donors (pink), hydrophobic regions (blue) and the aromatic rings in yellow are shown in the figure.
Protein backbone RMSD plots indicate the stability of the IPCS-inhibitor complex. The drug backbone RMSD plots indicate that compounds 2 and 3 maintained their interactions with the IPCS protein (Figure 4). Binding modes of compounds 1 to 5 post MD simulation have been shown in Figure 5a–e.
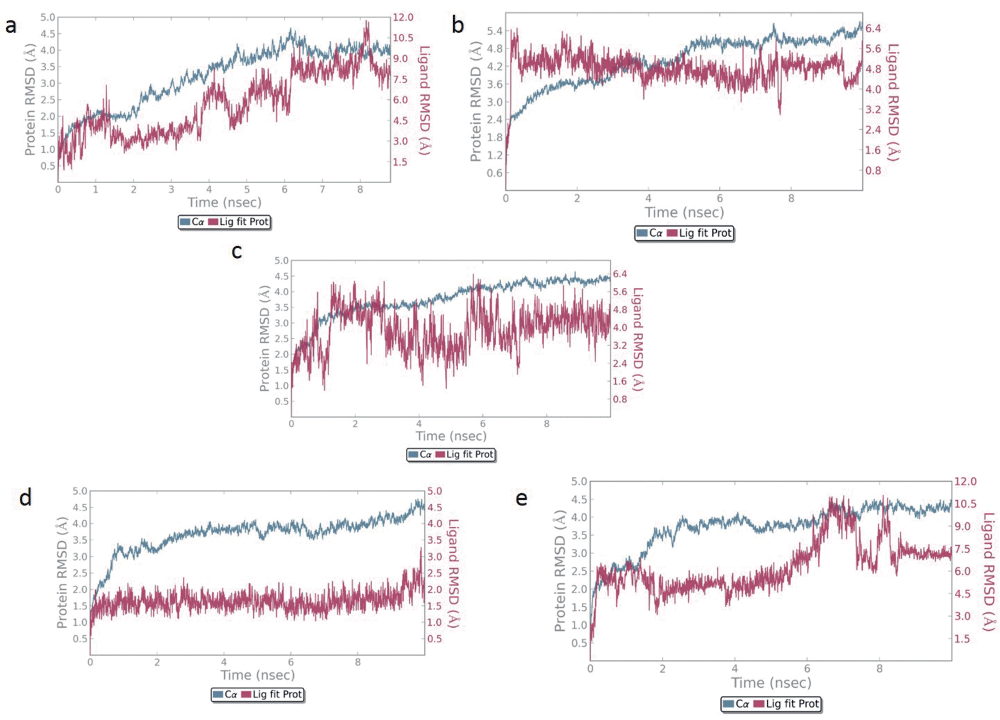
Backbone RMSD of a) Compound 1 and b) Compound 2 c) Compound 3 d) Compound 4 e) Compound 5 is shown in the figure. Compound 1, 2 and 3 appear to maintain their stability within the binding pocket as they show lower RMSD fluctuations.
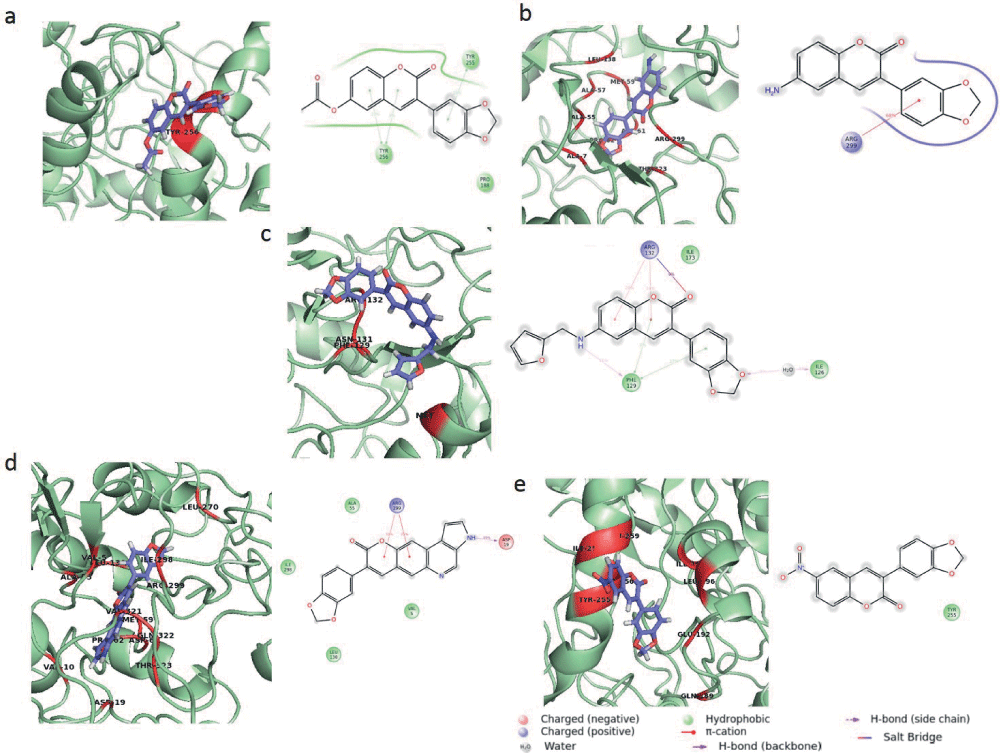
The interaction of the ligand within the IPCS inhibitor complex post MD simulation is shown the figure a) IPCS - compound 1 complex b) IPCS - compound 2 complex c) IPCS - compound 3 complex d) IPCS - compound 4 complex and e) IPCS - compound 5 complex. MD simulation was performed for a time period of 10ns. Interacting residues are represented in red.
IPCS (Inositol phosphorylceramide synthase) has been identified as an important drug target in the sphingolipid metabolism of several organisms like fungi, yeast and protozoans like Leishmania and Trypanosoma28. Systems biology has played a major role in defining the relative importance of IPCS in the sphingolipid metabolism of Leishmania, a protozoan responsible for causing an infectious disease leishmaniasis. The quest for developing new inhibitors for any target protein relies mainly on in silico approaches like computer based docking which involves the generation of a comprehensive set of ligand conformations that are eventually scored and ranked according to their stability and affinity for the protein. Coumarin has been shown to simulate the macrophages, enhancing their phagocytic ability29. A total of five ligands were developed as inhibitors for the IPCS protein. Molecular docking of the inhibitors with the IPCS protein revealed the binding modes of inhibitors. To account for the flexibility of the protein and ligand and to determine the binding affinity of the inhibitors with the IPCS protein, a 10 ns molecular dynamics simulation of the docked complexes was carried out. Binding mode analysis revealed that the binding modes obtained after MD simulation were more or less similar to that obtained post docking (Table 4). The presence of a large number of H bond acceptors, H bond donors as well as hydrophobic groups in the ligands account for the stability of the ligand inside the binding pocket of IPCS. Based on the RMSD of the ligand-protein complex, it was observed that compounds 1, 2 and 3 maintained their interaction with the protein with lower RMSD fluctuations. Out of these, compound 3 showed the highest binding affinity and its cytotoxicity was assessed using flow cytometry. Cytotoxicity of compound 3 was lesser as compared to other compound. A comparison of compound 3 treated macrophages along with the untreated macrophages has been made in Figure 6.

Macrophages were treated with compound 3 for 24h. a) Control cell population displayed a percentage viability of 73.5% b) Compound 3 (1mg/ml) treated macrophages displayed a viability of 67.3% post 24hr treatment.
There is an urgent need to design and develop novel anti-leishmanial compounds due to various problems associated with the current chemotherapeutics for the treatment of this disease. IPCS has been proposed to be a probable drug target in the sphingolipid pathway of Leishmania. We have designed a few novel coumarin derivatives using in silico approaches. MD simulation post docking studies reveal the interactions between the IPCS protein and ligands. Binding modes obtained after docking and after MD simulation reveal almost identical binding modes which is suggestive of the selectivity and selectivity of the ligand towards the active site of the IPCS protein.
F1000Research: Dataset 1. Raw data for ‘Molecular docking and molecular dynamics simulation study of inositol phosphorylceramide synthase – inhibitor complex in leishmaniasis: Insight into the structure based drug design’, 10.5256/f1000research.9151.d12833730
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)