Keywords
Gene expression signatures, breast cancer, chemotherapy resistance, hormone therapy, machine learning, support vector machine, random forest
This article is included in the Bioinformatics gateway.
This article is included in the Machine learning: life sciences collection.
Gene expression signatures, breast cancer, chemotherapy resistance, hormone therapy, machine learning, support vector machine, random forest
Current pharmacogenetic analysis of chemotherapy makes qualitative decisions about drug efficacy in patients (determination of good, intermediate or poor metabolizer phenotypes) based on variants present in genes involved in the transport, biotransformation, or disposition of a drug. We have applied a supervised ML approach to derive accurate gene signatures, based on the biochemically-guided response to chemotherapies with breast cancer cell lines1, which show variable responses to growth inhibition by paclitaxel and gemcitabine therapies2,3. We analyzed stable4 and linked unstable genes in pathways that determine their disposition. This involved investigating the correspondence between 50% growth inhibitory concentrations (GI50) of paclitaxel and gemcitabine and gene copy number, mutation, and expression first in breast cancer cell lines and then in patients1. Genes encoding direct targets of these drugs, metabolizing enzymes, transporters, and those previously associated with chemo-resistance to paclitaxel (n=31 genes) were then pruned by multiple factor analysis (MFA), which indicated expression of ABCC10, BCL2, BCL2L1, BIRC5, BMF, FGF2, FN1, MAP4, MAPT, NKFB2, SLCO1B3, TLR6, TMEM243, TWIST1, and CSAG2 could predict sensitivity in breast cancer cell lines with 84% accuracy. The cell line-based paclitaxel-gene signature predicted sensitivity in 84% of patients with no or minimal residual disease (n=56; data from 5). The present study derives related gene signatures with ML approaches that predict outcome of hormone- and chemotherapies in the large METABRIC breast cancer cohort6.
SVM learning: Previously, paclitaxel-related response genes were identified from peer-reviewed literature, and their expression and copy number in breast cancer cell lines were analyzed by multiple factor analysis of GI50 values of these lines2 (Figure 1). Genes with expression levels related to GI50 were used to derive SVMs by backwards feature selection for paclitaxel, tamoxifen, methotrexate, 5-fluorouracil, epirubicin, and doxorubicin (trained using the function fitcsvm in MATLAB R2014a7 and tested with either leave-one-out or 9 fold cross-validation). These SVMs were then assessed for their ability to predict patient outcomes based on available metadata (see Figure 1 and reference 1). Interactive prediction using normalized expression values as input is available at http://chemotherapy.cytognomix.com.
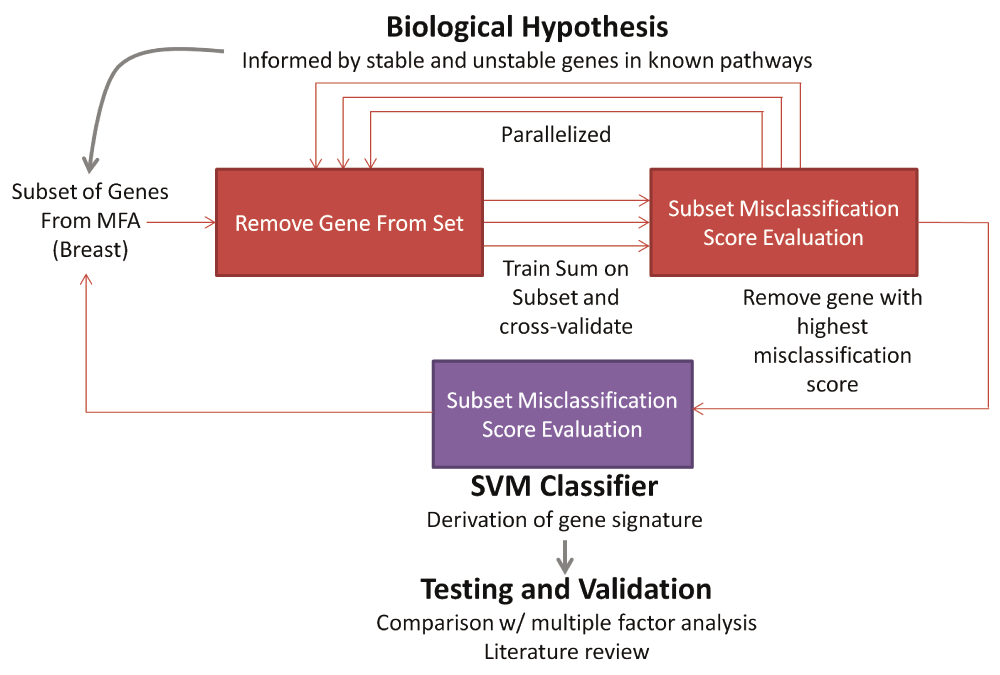
The initial set of genes is carefully selected through the understanding of the drug and the pathways associated with it. A multiple factor analysis of the GI50 values of a training set of breast cancer cell lines and the corresponding expression levels of each gene in the initial set reduces the list of genes. Given this expression levels of each gene the reduced set for each cell line, the method finds the optimal gene subset and the SVM that minimizes the misclassification rate by cross-validation. The SVM is evaluated on patients by classifying those with shorter survival time as resistant and longer survival as sensitive to hormone and/or chemotherapy. The Gaussian kernel SVM requires manual selection of two different parameters, C and sigma; these parameters determine how strictly the SVM learns the training set, thus if not selected properly can lead to overfitting. A grid search evaluates a wide range of combinations of these values by parallelization. The algorithm selects the C and sigma combination that lead to the lowest cross-validation misclassification rate. A backwards feature selection (greedy) algorithm is used, in which one gene of the set is left out in a reduced gene set and the classification is then assessed; genes that maintain or lower the misclassification rate are kept in the signature. The procedure is repeated until the subset with the lowest misclassification rate is selected as the optimal subset of genes.
RF learning: RF was trained using the WEKA 3.78 data mining tool. This classifier uses multiple random trees for classification, which are combined via a voting scheme to make a decision on the given input gene set. Figure 2 depicts the therapy outcome prediction process of a given patient using a RF consisting of a series of decision trees derived from different subsets of paclitaxel-related genes.
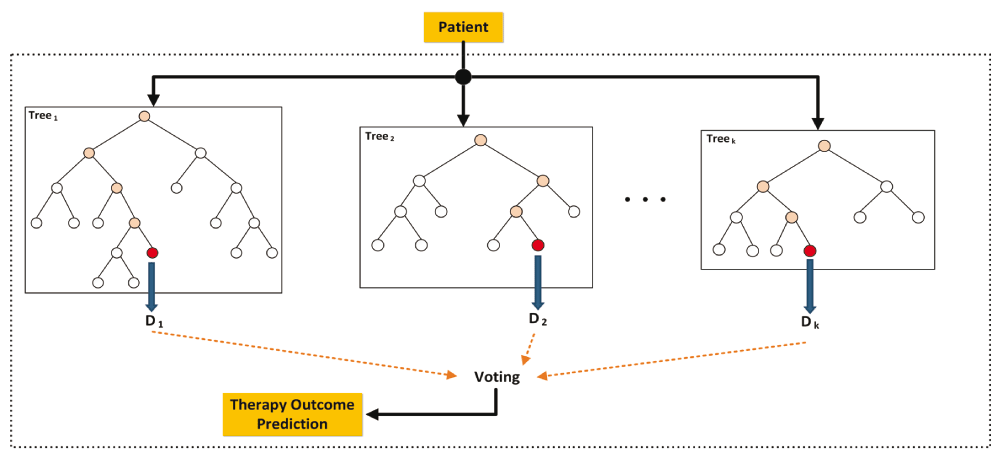
Several DTs are built using different subsets of paclitaxel-related genes. The process starts from the root of each tree and if the expression of the gene corresponding to that node is greater than a specific value, the process continues through the right branch, otherwise it continues through the left branch until it reaches a leaf node; that leaf represents the prediction of the tree for that specific input. The decisions of all trees are considered and the one with the largest number of votes is selected as the patient outcome.
Augmented Gene Selection: The most relevant genes (features) for therapy outcome prediction were found using the minimum redundancy and maximum relevance (mRMR) approach9. mRMR is a wrapper that incrementally selects genes by maximizing the average mutual information between gene expression features and classes, while minimizing their redundancies:
where fi corresponds to a feature in gene set S, I(fi,C) is the mutual information between fi and class C, and I(fi,fj) is the mutual information between features fi and fj.
For this experiment, we used a 26-gene signature (genes ABCB1, ABCB11, ABCC1, ABCC10, BAD, BBC3, BCL2, BCL2L1, BMF, CYP2C8, CYP3A4, MAP2, MAP4, MAPT, NR1I2, SLCO1B3, TUBB1, TUBB4A, TUBB4B, FGF2, FN1, GBP1, NFKB2, OPRK1, TLR6, TWIST1) as the base feature set. These genes were selected (in Ref. 1) based either on their known involvement in paclitaxel metabolism, or evidence that their expression levels and/or copy numbers correlate with paclitaxel GI50 values (Table 3). mRMR and SVM were combined to obtain a subset of genes that can accurately predict patient survival outcome; here, we considered 3, 4 and 5 years as survival thresholds for breast cancer patients (Table 3).
| Patient treatment | # of patients | Agent: final gene signature | Accuracy (%) |
|---|---|---|---|
| CT and CT/HT combination (without radiation therapy)1 | 84 | Paclitaxel: ABCC1, ABCC10, BAD, BIRC5, FN1, GBP1, MAPT, SLCO1B3, TMEM243, TUBB3, TUBB4B | 78.6 |
| Tamoxifen: ABCC2, ALB, CCNA2, E2F7, FLAD1, FMO1, NCOA2, NR1I2, PIAS4, SULT1E1 | 76.2 | ||
| Methotrexate: ABCC2, ABCG2, CDK2, DHFRL1 | 71.3 | ||
| Epirubicin: ABCB1, CDA, CYP1B1, ERBB3, ERCC1, MTHFR, PON1, SEMA4D, TFDP2 | 72.6 | ||
| Doxorubicin: ABCC2, ABCD3, CBR1, FTH1, GPX1, NCF4, RAC2, TXNRD1 | 75.0 | ||
| 5-Fluorouracil: ABCB1, ABCC3, MTHFR, TP53 | 71.4 | ||
| CT and/or HT1,2,3,4 | 735 | Paclitaxel: BAD, BCAP29, BCL2, BMF, CNGA3, CYP2C8, CYP3A4, FGF2, FN1, NFKB2, NR1I2, OPRK1, SLCO1B3, TLR6, TUBB1, TUBB3, TUBB4A, TUBB4B, TWIST1 | 66.1 |
| Deceased only2,4,5 (CT and/or HT) | 327 | Paclitaxel: ABCB11, BAD, BBC3, BCL2, BCL2L1, BIRC5, CYP2C8, FGF2, FN1, GBP1, MAPT, NFKB2, OPRK1, SLCO1B3, TMEM243 | 75.2 |
| No treatment1 | 304 | Paclitaxel: ABCB1, ABCB11, BBC3, BCL2L1, BMF, CYP3A4, FGF2, GBP1, MAP4, MAPT, NR1I2, OPRK1, SLCO1B3, TUBB4A, TUBB4B, TWIST2 | 73.4 |
Initial gene sets preceding feature selection: Paclitaxel - ABCB1, ABCB11, ABCC1, ABCC10, BAD, BBC3, BCAP29, BCL2, BCL2L1, BIRC5, BMF, CNGA3, CYP2C8, CYP3A4, FGF2, FN1, GBP1, MAP2, MAP4, MAPT, NFKB2, NR1I2, OPRK1, SLCO1B3, TLR6, TUBB1, TWIST1. Tamoxifen - ABCB1, ABCC2, ALB, C10ORF11, CCNA2, CYP3A4, E2F7, F5, FLAD1, FMO1, IGF1, IGFBP3, IRS2, NCOA2, NR1H4, NR1I2, PIAS4, PPARA, PROC, RXRA, SMARCD3, SULT1B1, SULT1E1, SULT2A1. Methotrexate - ABCB1, ABCC2, ABCG2, CDK18, CDK2, CDK6, CDK8, CENPA, DHFRL1. Epirubicin - ABCB1, CDA, CYP1B1, ERBB3, ERCC1, GSTP1, MTHFR, NOS3, ODC1, PON1, RAD50, SEMA4D, TFDP2. Doxorubicin - ABCB1, ABCC2, ABCD3, AKR1B1, AKR1C1, CBR1, CYBA, FTH1, FTL, GPX1, MT2A, NCF4, RAC2, SLC22A16, TXNRD1. 5-Fluorouracil - ABCB1, ABCC3, CFLAR, IL6, MTHFR, TP53, UCK2.
1 Surviving patients; 2 Analysis included patients in the METABRIC ‘discovery’ dataset only; 3 SVMs tested with 9 fold cross-validation, all others tested with leave-one-out cross-validation; 4 Includes all patients treated with HT,CT, combination CT/HT, either with or without combination radiotherapy; 5 Median time after treatment until death (> 4.4 years) was used to distinguish favorable outcome, ie. sensitivity to therapy.
| Type of treatment | Survival years (as threshold) | # Patients | Final gene expression signature | Accuracy (%) | AUC1 |
|---|---|---|---|---|---|
| Chemotherapy (CT) | 3 | 53 | ABCB1, ABCB11, ABCC1, ABCC10, BAD, BBC3, BCL2, BCL2L1, BMF, CYP2C8, CYP3A4, MAP2, MAP4, MAPT, NR1I2, SLCO1B3, TUBB1, TUBB4A, TUBB4B | 56.6 | 0.534 |
| 4 | 60.4 | 0.645 | |||
| 5 | 58.5 | 0.645 | |||
| Hormone therapy (HT) | 3 | 420 | 82.4 | 0.641 | |
| 4 | 77.4 | 0.555 | |||
| 5 | 70.7 | 0.648 | |||
| CT and/or HT | 3 | 504 | 79.6 | 0.564 | |
| 4 | 73.6 | 0.527 | |||
| 5 | 63.5 | 0.563 |
| Type of treatment | Survival years (as threshold) | Number of patients1 | Gene signature selected by mRMR | Accuracy (%) | AUC |
|---|---|---|---|---|---|
| Chemotherapy (CT) | 3 | 53 | BCL2 , TWIST1 | 73.6 | 0.724 |
| 4 | ABCB11, ABCC1, BAD, BBC3, BCL2L1 | 79.2 | 0.793 | ||
| 5 | ABCB11, BAD, CYP2C8, CYP3A4, MAP2, MAPT, FGF2 | 77.4 | 0.759 | ||
| Hormone therapy (HT) | 3 | 420 | ABCB11, ABCC1, BAD, BCL2, CYP2C8, CYP3A4, MAP4, MAPT, NR1I2, TUBB1, GBP1, OPRK1 | 84.0 | 0.532 |
| 4 | BBC3, MAP4, FGF2, OPRK1 | 79.3 | 0.519 | ||
| 5 | ABCC10, MAPT, TUBB1 | 72.6 | 0.520 | ||
| CT and/or HT | 3 | 504 | ABCB11, ABCC1, BBC3, BCL2, BCL2L1, CYP2C8, CYP3A4, MAP2, MAPT, TLR6, TWIST1 | 74.6 | 0.565 |
| 4 | ABCB11, BAD, BCL2, CYP3A4, MAP2, MAP4, NR1I2, OPRK1, TWIST1 | 75.0 | 0.535 | ||
| 5 | TWIST1, BCL2, BMF, CYP2C8, CYP3A4, BCL2L1, BBC3, TLR6, BAD, MAP4, NR1I2, GBP1, NFKB2 | 65.9 | 0.525 |
1Predicted treatment responses for individual METABRIC patients using the described ML techniques are provided in Dataset 1.
The performance of several ML techniques have been compared that distinguish paclitaxel sensitivity and resistance in METABRIC patients using its tumour gene expression datasets. SVMs have generated gene signatures, indicating which genes are important for treatment response in METABRIC patients. These models are more accurate for prediction of outcomes in patients receiving HT and/or CT compared to other patient groups.
SVMs and RF were trained using expression of genes associated with paclitaxel response, mechanism of action and stable genes in the biological pathways of these targets (Figure 3). SVM models for drugs used to treat these patients were derived by backwards feature selection on patient subsets stratified by treatment or outcome (Table 1). The highest SVM accuracy was found for the paclitaxel signature in patients treated with HT and/or adjuvant chemotherapy (78.6%).
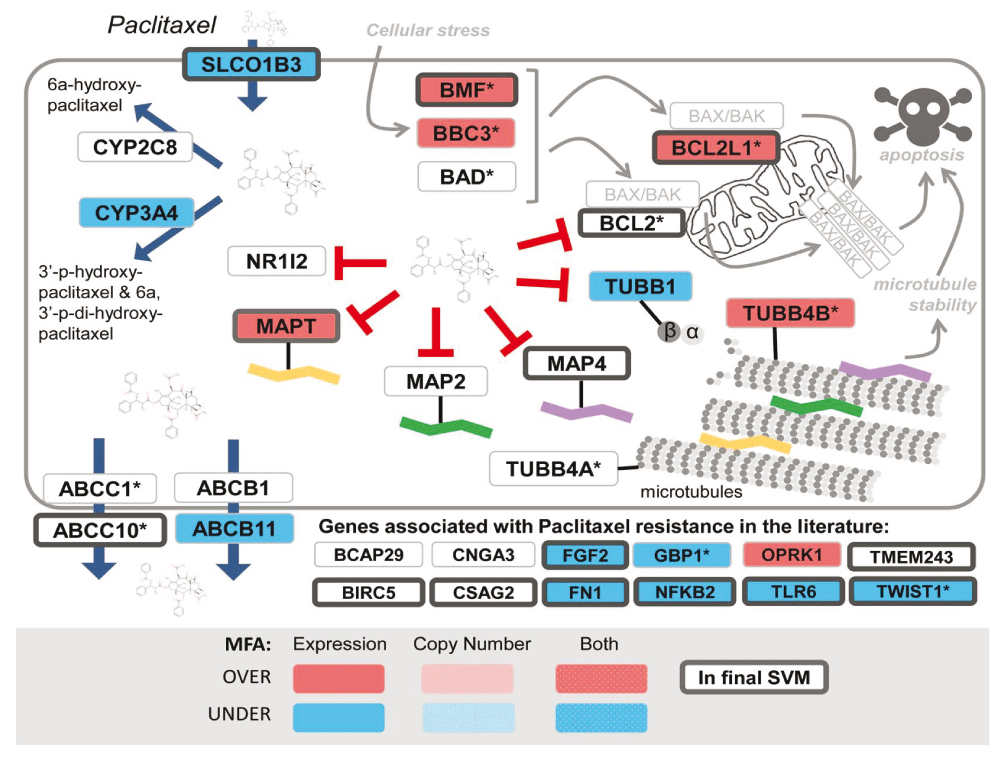
Schematic elements of gene expression changes associated with response to paclitaxel. Red boxes indicate genes with a positive correlation between gene expression or copy number, and resistance using multiple factor analysis. Blue demonstrates a negative correlation. Genes outlined in dark grey are those in a previously published paclitaxel SVM model (reproduced from reference 1 with permission).
The RF classifier was used to predict paclitaxel therapy outcome for patients that underwent CT and/or HT (Table 2). The best performance achieved with RF showed 82.4% overall accuracy using a 3-year survival threshold for distinguishing therapeutic resistance vs. sensitivity.
The best overall accuracy and AUC (sensitivity and specificity) for CT/HT patients using mRMR feature selection for SVM predicting outcome of paclitaxel therapy was obtained for CT patients with 4 year survival. Outcomes for HT patients with 3 year survival were predicted with 84% accuracy; however the specificity was lower in this group. SVM combined with mRMR further improved accuracy of feature selection and prediction of response to hormone and/or chemotherapy based on survival time than either SVM or RF alone.
While not a replication study sensu stricto, the initial paclitaxel gene set used for feature selection was the same as in our previous study1. Predictions for the METABRIC patient cohort, which was independent of the previous validation set5, of the either same (SVM) or different ML methods (RF and SVM with mRMR) exhibited comparable or better accuracies than our previous gene signature1.
These techniques are powerful tools which can be used to identify genes that may be involved in drug resistance, as well as predict patient survival after treatment. Future efforts to expand these models to other drugs may assist in suggesting preferred treatments in specific patients, with the potential impact of improving efficacy and reducing duration of therapy.
Patient data: The METABRIC datasets are accessible from the European Genome-Phenome Archive (EGA) using the accession number EGAS00000000083 (https://www.ebi.ac.uk/ega/studies/EGAS00000000083). Normalized patient expression data for the discovery (EGAD00010000210) and validation sets (EGAD00010000211) were retrieved with permission from EGA. Corresponding clinical data was obtained from the literature6. While not individually curated, HT patients were treated with tamoxifen and/or aromatase inhibitors, while CT patients were most commonly treated with cyclophosphamide-methotrexate-fluorouracil (CMF), epirubicin-CMF, or doxorubicin-cyclophosphamide.
F1000Research: Dataset 1. Predicted treatment response for each individual METABRIC patient, 10.5256/f1000research.9417.d13398310
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)